
Viroma se refiere al conjunto de virus que a menudo se investiga y describe mediante la secuenciación metagenómica de ácidos nucleicos virales que se encuentran asociados con un ecosistema, organismo u holobionte en particular. La palabra se usa con frecuencia para describir los metagenomas virales de escopeta ambientales. Los virus, incluidos los bacteriófagos, se encuentran en todos los entornos, y los estudios del viroma han proporcionado información sobre el ciclo de los nutrientes, desarrollo de la inmunidad, y una fuente importante de genes a través de la conversión lisogénica.
Historia
Los primeros estudios exhaustivos de viromas se realizaron mediante secuenciación comunitaria de escopeta, que con frecuencia se denomina metagenómica. En la década de 2000, el laboratorio Rohwer secuenció viromas de agua de mar, sedimentos marinos, heces humanas adultas, heces humanas infantiles, tierra, y sangre. Este grupo también realizó el primer viroma de ARN con colaboradores del Instituto Genómico de Singapur. A partir de estos primeros trabajos, se concluyó que la mayor parte de la diversidad genómica está contenida en el viroma global y que la mayor parte de esta diversidad permanece sin caracterizar. Este punto de vista fue apoyado por el proyecto de secuenciación genómica individual, en particular el fago de mycobacterium.
Métodos de estudio
Para estudiar el viroma, las partículas similares a virus se separan de los componentes celulares, generalmente mediante una combinación de filtración, centrifugación de densidad y tratamientos enzimáticos para eliminar los ácidos nucleicos libres. A continuación, los ácidos nucleicos se secuencian y analizan utilizando métodos metagenómicos . Alternativamente, existen métodos computacionales recientes que utilizan secuencias ensambladas directamente metagenómicas para descubrir virus.
Global Ocean Viromes (GOV) es un conjunto de datos que consiste en una secuenciación profunda de más de 150 muestras recolectadas en los océanos del mundo en dos períodos de estudio por un equipo internacional.
Anfitriones de virus
Los virus son las entidades biológicas más abundantes en la Tierra, pero los desafíos para detectar, aislar y clasificar virus desconocidos han impedido realizar estudios exhaustivos del viroma global. Se utilizaron más de 5 Tb de datos de secuencia metagenómica de 3.042 muestras geográficamente diversas para evaluar la distribución global, la diversidad filogenética y la especificidad del hospedador de los virus.

En agosto de 2016, más de 125.000 genomas virales de ADN parcial, incluido el fago más grande identificado hasta ahora, aumentaron 16 veces el número de genes virales conocidos. Se utilizó un conjunto de métodos computacionales para identificar las conexiones putativas del virus huésped. La información del hospedador viral aislado se proyectó en un grupo, lo que resultó en asignaciones de hospedador para el 2,4% de los grupos virales.
Luego, el sistema inmunológico procariótico CRISPR-Cas que contiene una "biblioteca" de fragmentos del genoma de los fagos (proto-espaciadores) que han infectado previamente al huésped. Se identificaron espaciadores de genomas microbianos aislados con coincidencias con contigs virales metagenómicos (mVC) para el 4,4% de los grupos virales y el 1,7% de los únicos. Se exploró la hipótesis de que los genes de ARN de transferencia viral (ARNt) se originan en su anfitrión.
Los ARNt virales identificados en el 7,6% de las mVC se emparejaron para aislar genomas de una sola especie o género. La especificidad de la asignación viral del hospedador basada en ARNt fue confirmada por coincidencias de espaciador CRISPR-Cas que muestran una concordancia del 94% en el ámbito de género. Estos enfoques identificaron 9.992 asociaciones putativas de virus-huésped que permiten la asignación de huéspedes al 7,7% de las mVC. La mayoría de estas conexiones eran previamente desconocidas e incluyen huéspedes de 16 filos procariotas para los que no se han identificado virus previamente.
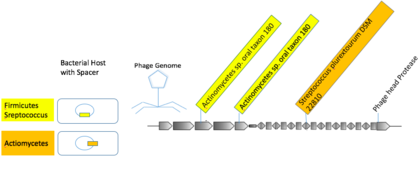
Muchos virus se especializan en infectar hospedadores relacionados. Es posible que existan generalistas virales que infectan a huéspedes de diferentes órdenes taxonómicos. La mayoría de las coincidencias de espaciadores CRISPR fueron de secuencias virales a huéspedes dentro de una especie o género. Algunos mVC se vincularon a múltiples huéspedes de taxones superiores. Un grupo viral compuesto por macs de muestras orales humanas contenía tres foto-espaciadores distintos con coincidencias casi exactas con los espaciadores en Actionbacteria y Firmicutes.
Para enero de 2017, el sistema IMG/VR, la base de datos de virus pública interactiva más grande, contenía 265.000 secuencias virales metagenómicas y virus aislados. Este número aumentó a más de 760.000 en noviembre de 2018 (IMG/VR v.2.0). Los sistemas IMG/VR sirven como punto de partida para el análisis de secuencia de fragmentos virales derivados de muestras metagenómicas.
| Control de autoridades |
|
|---|
-
 Datos: Q24977348
Datos: Q24977348