
| Enfermedad de Tay-Sachs | ||
|---|---|---|
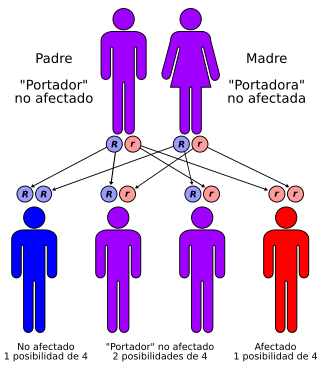 Esquema genético.
| ||
| Especialidad |
pediatría neurología genética médica |
|
| Sinónimos | ||
|
Sipoidosis infantil GM 2 gangliosidosis, tipo S. Idiocia amaurótica familiar infantil. Gangliosidosis cerebral con degeneración infantil cerebromacular. Lipidosis por gangliósidos infantil. Esfingolipidosis de Tay Sachs. Idiocia amaurótica familiar. | ||
La enfermedad de Tay-Sachs es una enfermedad rara genética que afecta al cerebro y al sistema nervioso central y es de carácter hereditario, autosómico recesivo.
Se trata de una de las enfermedades por depósito lisosomal. Los individuos que la padecen son incapaces de producir una enzima lisosómica llamada hexosaminidasa-A que participa en la degradación de los gangliósidos, un tipo de esfingolípido, que se acumulan y degeneran al sistema nervioso central. Se incluye dentro de las lipidosis o enfermedades por almacenamiento de lípidos.
Fisiopatología
La enfermedad de Tay-Sachs es una anomalía autosómica recesiva que da lugar a una degeneración progresiva del sistema nervioso central. Los bebés parecen normales al nacer y se desarrollan normalmente hasta los seis meses, perdiendo luego gradualmente sus capacidades físicas y mentales. Los bebés afectados quedan ciegos, sordos, mentalmente retrasados y paralizados solo en uno o dos años y la mayoría no viven más allá de los cinco años. Recibe el nombre de los primeros investigadores que describieron los síntomas y los relacionaron con la enfermedad hacia finales del siglo XIX, Warren Tay y Bernard Sachs.
Etiología
La enfermedad de Tay-Sachs se produce como consecuencia de la pérdida de actividad de la enzima hexosaminidasa A (Hex-A). Esta enzima se encuentra normalmente en los lisosomas, orgánulos que degradan moléculas grandes para reciclarlas para la célula. La Hex-A se necesita para degradar el gangliósido GM2, un componente lipídico de las membranas de las células nerviosas. Sin Hex-A funcional, los gangliósidos se acumulan en las células cerebrales y dan lugar al deterioro del sistema nervioso. Los portadores heterocigotos para TSD, con una copia normal del gen, producen solo la mitad de la cantidad normal de Hex-A, pero no manifiestan síntomas de la enfermedad.
El gen responsable de la enfermedad de Tay-Sachs se encuentra en el cromosoma 15 y codifica para la subunidad alfa de la enzima Hex-A. Desde que se aisló el gen en 1985, se han identificado más de 50 mutaciones distintas que dan lugar a la TSD. Aunque la forma más corriente de la enfermedad es la infantil, en donde no se produce Hex-A funcional, hay también una forma rara de aparición tardía que se da en pacientes con una actividad muy reducida de la Hex-A. La TSD de aparición tardía no es detectable hasta que los pacientes tienen veinte o treinta años y en general es mucho menos grave que la forma infantil.
Cuadro clínico
Entre los síntomas más habituales están el temblor de las manos, defectos del habla, debilidad muscular y pérdida del equilibrio, así como sordera, pérdida de la capacidad visual, incluso llegando a la ceguera, crisis epilépticas, retraso del crecimiento, irritabilidad, apatía y retrasos de las capacidades mentales y sociales.
Pronóstico
Esta es una enfermedad incurable, los pacientes generalmente fallecen antes de los cinco años de vida. Se reportó un caso de un paciente que vivió hasta los 16 años
Tratamiento
Aunque actualmente no hay tratamientos efectivos para la TSD, avances recientes en la detección de portadores ha ayudado a reducir la prevalencia de la enfermedad en poblaciones de alto riesgo. Los portadores se pueden identificar mediante pruebas de la actividad de la Hex-A o mediante pruebas de ADN que detecten mutaciones génicas concretas. Además, actualmente se están investigando inhibidores de la síntesis de gangliósidos y terapias de sustitución de la enzima Hex-A como tratamientos potenciales para la enfermedad de Tay-Sachs.
Los últimos avances en ingeniería genética permiten incluir en el ADN del niño, ya sintomático de su carencia, el gen reparador de la anomalía causada por la falta de producción de la enzima Hex-A. Esta compleja técnica se realiza mediante la introducción del gen previamente incluido en un virus de muy baja actividad (mucho menor que la del virus del resfrío común) al que se le han dejado los genes capaces de generar la rápida diseminación corporal de dicho virus y eliminado los genes patógenos que normalmente poseía.
Prevención
Hoy en día existen diferentes estrategias de prevención de la enfermedad de Tay-Sachs, orientadas principalmente a las poblaciones judías, las más afectadas por esta enfermedad:
Diagnóstico prenatal
La prueba genética prenatal puede determinar si el feto ha heredado una copia defectuosa del gen de ambos padres. Para las parejas que están dispuestos a interrumpir el embarazo, esto elimina el riesgo de enfermedad de Tay-Sachs, pero el aborto plantea problemas éticos para muchas familias. La muestra de vellosidad coriónica, que se puede realizar después de la semana 10 de gestación, es la forma más común de diagnóstico prenatal.
Diagnóstico genético preimplantacional
En los casos de fecundación, en donde se extraen óvulos de la madre para su fecundación, se puede observar el embrión antes de ser implantado en el útero de la madre, seleccionándose así solo los embriones sanos. Además de la enfermedad de Tay-Sachs, el diagnóstico genético preimplantacional se ha utilizado para prevenir la fibrosis quística, anemia de células falciformes, la enfermedad de Huntington y otros trastornos genéticos. Sin embargo, este método es costoso. Requiere tecnologías médicas invasivas y está más allá de los recursos financieros de muchos.
Elección de parejas
En los círculos de judíos Ashkenazis se llevan a cabo estudios para determinar qué personas de la comunidad son portadoras del gen y por tanto pueden transmitir a sus hijos la enfermedad. Estos resultados no se les dan directamente a las personas, sino que se les facilita un número de 6 dígitos al que pueden llamar para conocer si son portadores o no. El objetivo de estas pruebas es evitar que 2 personas portadoras tengan hijos entre ellos.
Principales lipidosis
- Enfermedad de Gaucher.
- Enfermedad de Niemann-Pick.
- Enfermedad de Fabry
- Enfermedad de Wolman
- Xantomatosis cerebrotendinosa
- Sitosterolemia
- Enfermedad de Refsum
- Enfermedad de Tay-Sachs
- Leucodistrofia metacromática.
- William S. Klug "Conceptos de Genética", 8ª Edición.
Enlaces externos
- Instituto Carlos III
- The National Tay-Sachs and Allied Diseases Association Inc. (en inglés)
- Catálogo McKusick (en inglés)
| Control de autoridades |
|
|---|
-
 Datos: Q560337
Datos: Q560337
-
 Multimedia: Tay–Sachs disease / Q560337
Multimedia: Tay–Sachs disease / Q560337