
| Enfermedad de Lafora | ||
|---|---|---|
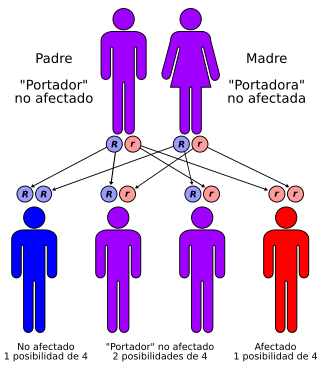 La enfermedad de Lafora tiene un patrón de herencia autosómica recesiva
| ||
| Especialidad | neurología | |
| Sinónimos | ||
| Síndrome de Lafora, Enfermedad de cuerpos de Lafora, Epilepsia mioclónica de Lafora,Epilepsia mioclónica progresiva tipo 2 (EPM2) | ||
La enfermedad de Lafora o síndrome de Lafora es una forma de epilepsia mioclónica progresiva que se presenta generalmente en países del sur de Europa. En España, se han presentado 20-30 casos. Se caracteriza por mioclonos, convulsiones epilépticas y demencia (una pérdida progresiva de la memoria y de otras funciones intelectuales).
Fue descrita en 1911 por el neuropatólogo español Gonzalo Rodríguez Lafora.
Evolución natural de la enfermedad
Los primeros signos clínicos aparecen en la pubertad y adolescencia (6-20 años) y en principio se manifiestan como crisis epilépticas convulsivas o crisis visuales, que suelen describirse como visión de luces o estrellas. Poco después aparecen las mioclonías (sacudidas involuntarias de los brazos y piernas). Durante el transcurso de la enfermedad se producen alteraciones en la marcha, ceguera y afectación de los músculos y de los nervios.
Su evolución está marcada por una degeneración progresiva del sistema nervioso y por un deterioro de las funciones cerebrales, conduciendo a un estado de dependencia total. El enfermo se vuelve incapaz de moverse, de hablar, de alimentarse solo, etc. Los enfermos fallecen alrededor de los 10 años después de la aparición de los primeros signos neurológicos.
Es una enfermedad hereditaria transmitida de un modo autosómico recesivo, lo que quiere decir que la enfermedad ocurre solamente cuando se hereda dos copias del gen defectuoso, uno de cada padre. No tiene predilección por ningún sexo.
Causas
La deficiencia de laforina (EPM2A) es la causa de la epilepsia mioclónica progresiva tipo 2 (EPM2), también conocida como enfermedad de Lafora.
La laforina (EPM2A) es una proteína tipo fosfatasa dual específica que participa en el control del metabolismo del glicógeno, particularmente en la monitorización y prevención de la formación de moléculas de glicógeno poco ramificadas. Interacciona consigo misma y también con PPP1R5, HIRIP5 y EPM2AIP1. Se une al glicógeno y a los cuerpos de Lafora.
Cuerpos de Lafora
A nivel celular, la enfermedad de Lafora se caracteriza por una acumulación de poliglucosanos parecidos al almidón, llamados cuerpos (o corpúsculos) de Lafora, que son más abundantes en los órganos con un metabolismo de glucosa más alto: cerebro, corazón, hígado y músculo esquelético. Entre otras condiciones que involucran a los poliglucosanos, la laforina es única en el sentido que las inclusiones están en las dendritas neuronales pero no en los axones y los poliglucosanos formados están asociados con el retículo endoplasmático.
Signo de Lafora
Cosquilleo de la nariz, considerado como signo precoz de meningitis cerebroespinal. No es raro en la fase precoz de la meningitis tuberculosa; se localiza, muchas veces, en la nariz, obligando a rascarse violentamente a los enfermos, sobre todo a los niños, hasta hacerse sangre.
Epónimo
La enfermedad lleva el nombre de Gonzalo Rodríguez Lafora, neuropatólogo español que reconoció, por primera vez en 1911, los pequeños cuerpos de inclusión en quienes presentan la enfermedad que lleva su nombre.