
| Enfermedad de Gaucher | ||
|---|---|---|
 | ||
| Especialidad |
endocrinología neurología |
|
| Sinónimos | ||
|
Síndrome de lipidosis cerebral Lipoidosis de querasina Lipidosis por glucosil cerebrósido | ||
La enfermedad de Gaucher (GD) es una enfermedad congénita autosómica recesiva, provocada por un déficit de la enzima glucocerebrosidasa (participante en la degradación lisosómica de los glucolípidos), lo que conlleva un acúmulo de glucocerebrósido (un tipo de esfingolípido) en las células del sistema fagocítico mononuclear. Está considerada como una "enfermedad rara". Se incluye dentro del grupo de las lipidosis que son producidas por acumulación de lípidos y pertenecen al más amplio grupo de las enfermedades por depósito lisosomal. Los síntomas principales consisten en alteraciones hemáticas, hepatomegalia y esplenomegalia.
Las células que acumulan el glucocerebrósido son denominadas células de Gaucher, y adquieren un aspecto característico: se muestran grandes, con aspecto mesenquimatoso, núcleo no desplazado y citoplasma con aspecto de "celofán arrugado". Las podemos ver sobre todo en médula ósea, hígado, bazo y ganglios linfáticos.
Las manifestaciones de la enfermedad de Gaucher incluyen anemia, trombocitopenia, hepatoesplenomegalia y lesiones óseas. Más del 90 % de los pacientes sufren trastornos óseos. El malestar, el dolor característico y la incapacidad producidos por estos trastornos óseos afectan gravemente a la calidad de vida de quienes la padecen.
Se puede manifestar en cualquier momento de la vida: niños, jóvenes, adultos o ancianos. Es importante realizar el diagnóstico precoz e instaurar tratamiento lo antes posible, para lograr la mejoría de los síntomas y las deficiencias que presenta el paciente.
Las técnicas principales para su diagnóstico son el ensayo fluorométrico de la actividad de la enzima responsable, la búsqueda de macrófagos que tienen el citoplasma con aspecto de "celofán arrugado" y el núcleo excéntrico en la médula ósea conocidas como "células Gaucher". También se realizan estudios genéticos, pero estos son menos comunes, pues se han identificado 200 mutaciones diferentes del gen que codifica la glucocerebrosidasa. Este se basa en el estudio de 4 mutaciones comunes y otras 7 menos frecuentes.
Historia
La enfermedad de Gaucher recibe su nombre en honor del médico francés Philippe Charles Ernest Gaucher, que realizó por primera vez la descripción del trastorno en el año 1882, tras observar a un paciente que presentaba aumento del tamaño del hígado y bazo (hepatoesplenomegalia). La Enfermedad del Gaucher surge en el año de 1882 en dónde el francés Philipe Gaucher describió el caso de una mujer joven que presentaba agrandamiento del bazo y células congestionadas características de la enfermedad, pero sin encontrar el origen bioquímico del trastorno.
En 1924 se empezaba a estudiar el caso más a fondo, en donde el médico alemán H. Lieb comenzó a indagar en la búsqueda de la razón de este caso, aislando un particular compuesto graso del bazo.
En 1932, las investigaciones de Aghion identificaron el compuesto graso aislado de los pacientes con esta enfermedad, como glucocerebrósido, que es un componente de las membranas celulares de los glóbulos rojos y blancos.
En 1948 Groen JF fue quien descubrió que la enfermedad tenía transmisión genética, localizando en el cromosoma 1 la anomalía que hacía que esa enfermedad sea hereditaria. Además, de comprobar que en cualquier raza se puede heredar la enfermedad, siempre y cuando se encuentre la anomalía en el cromosoma 1.
En 1965 Brady Roscoe descubrió la causa de la EG, el déficit de la enzima lisosómica betaglucosidasa ácida, la enzima es la que ayuda a degradar los glucolípidos del cuerpo, pero al tener escasez de esta, los glucolípidos empiezan a acumularse de manera saturada en el cuerpo, inclusive en el área del cerebro, derivando complicaciones y deterioro del paciente. Estas investigaciones fundamentaron las bases de la investigación de la terapia de reemplazo de enzimas.
Hoy en día en la actualidad, a partir de 2009 se documentan casos en donde se aplica la terapia de reemplazo de enzimas, dando resultados positivos en pacientes con gestación de a partir de 15 semanas, logrando un parto reducido en complicaciones, así como un menor deterioro del paciente durante su desarrollo 9 meses post parto. En 1991 se creó el registro Internacional de Gaucher y en 1992 se incorporaron los primeros pacientes en Latinoamérica.
En el año 2008 se creó el grupo latinoamericano para la enfermedad de Gaucher (GLAEG) cuyos principales objetivos son fomentar la realización de consejos regionales, difundir el ingreso de pacientes al registro internacional y aumentar el conocimiento sobre la enfermedad para lograr mejorar la atención y la calidad de vida de los pacientes.
Hasta abril de 2010 ingresaron 5828 pacientes de todo el mundo, 911 (15,6 %) son de Latinoamérica. Este es el primer informe global de la enfermedad en la Región: hay un predominio del sexo femenino, la forma clínica más frecuente es el tipo I (95 %); al diagnóstico la mayoría son <20 años (68 %). Las manifestaciones clínicas más frecuentes al diagnóstico son esplenomegalia (96 %) y anemia (49 %), el 80 % presentó hallazgos radiológicos de compromiso óseo. En nuestra región, la gran mayoría de los pacientes (89 %) ha recibido alguna vez terapia de reemplazo enzimática con imiglucorasa, logrando, con un seguimiento prolongado (hasta 10 años), las metas terapéuticas que muestran la gran eficacia de la terapia. Si bien el porcentaje de pacientes con terapia es alto, las suspensiones de tratamiento son frecuentes. Las principales deficiencias son: la carencia de evaluaciones viscerales volumétricas, de densitometría y de estudios moleculares en algunos pacientes. El principal problema es el subdiagnóstico.
Características
La identificación de los tres principales tipos clínicos (1, 2 y 3) y otros dos subtipos (perinatal letal y cardiovascular) es útil para determinar el pronóstico y la gestión de la enfermedad. GD tipo 1 se caracteriza por la presencia de signos clínicos o radiológicos de enfermedad de los huesos (osteopenia, las lesiones focales líticas o escleróticas, y osteonecrosis), hepatoesplenomegalia, anemia y trombocitopenia, enfermedad pulmonar, y la ausencia de enfermedad primaria del sistema nervioso central. GD tipos 2 y 3 se caracterizan por ser una enfermedad neurológica primaria, en el pasado se distinguían por la edad de inicio y la tasa de progresión de la enfermedad, pero estas comparaciones no son absolutas y fueron desechadas.
Es una enfermedad con inicio antes de los dos años de edad del individuo, caracterizado por un desarrollo psicomotor limitado, y por la muerte a la edad de dos a cuatro años para la enfermedad tipo 2. Las personas con la enfermedad de Gaucher tipo 3 pueden presentar el inicio de los síntomas antes de los dos años, pero a menudo tienen un curso más lento y progresivo y pueden vivir hasta la tercera o cuarta década. La forma perinatal letal se asocia con alteraciones o ictiosiforme colodión de la piel o con hidropesía fetal no inmune. La forma cardiovascular se caracteriza por la calcificación de las válvulas aórtica y mitral, esplenomegalia leve, opacidades de la córnea, y oftalmoplejía supranuclear. También se han descrito complicaciones cardiopulmonares en todos los subtipos clínicos, aunque varían en frecuencia y gravedad, esto se puede deber a la edad o tiempo desde que empezó a desarrollarse esta enfermedad
Herencia

La enfermedad de Gaucher se hereda de forma autosómica recesiva. Está causada por genes mutantes y defectuosos que son transmitidos por los 2 progenitores del individuo afectado. Sus signos y sus síntomas pueden manifestarse a cualquier edad, aunque el Tipo II y III son más comúnmente diagnosticados en la niñez. Afecta a personas de cualquier grupo étnico o raza, sin embargo la enfermedad de Gaucher Tipo I es más común entre los judíos de ascendencia Ashkenazi (Europa Oriental). Dentro de este grupo su frecuencia es muy superior a la general.
Tipos
Existen tres tipos de presentación de la enfermedad: Tipo I, Tipo II y Tipo III.
- Tipo I. No neuropática. Es la forma más común y afecta a 1 de cada 40 000 a 60 000 niños nacidos vivos. La enfermedad Tipo I, no afecta el cerebro o al sistema nervioso central, inclusive se ha visto que algunos pacientes con la enfermedad de Gaucher Tipo I carecen de síntomas representativos, mientras que otros desarrollan síntomas severos que pueden ser fatales.
- Tipo II. Neuropática aguda. La enfermedad de Gaucher Tipo II es más rara y poco frecuente por eso se presenta en menos de 1 en 100 000 niños nacidos vivos. Sin embargo, las personas con EG Tipo II sufren efectos más severos que las que presentan EG Tipo I, debido a que pueden sufrir grandes problemas neurológicos en conjunto con la anemia, trombocitopenia y fuertes dolores por las crisis óseas características de la enfermedad, ocasionando que muchos de ellos no vivan pasados los dos años de edad.
- Tipo III. Neuropática crónica. El Tipo III es también póco común y afecta a menos de 1 en 100 000 niños nacidos vivos. Esta forma de la enfermedad también puede causar signos y síntomas neurológicos, pero estos son menos severos que los de la EG Tipo II. Los signos y síntomas aparecen durante la infancia o niñez, y los pacientes con la enfermedad de Gaucher Tipo III pueden vivir pasada la edad adulta.
Signos y síntomas
- Hepatomegalia y esplenomegalia indoloras; el tamaño del bazo puede llegar a los 3000 ml (siendo el tamaño normal de 50-200 ml), debido a la rápida y prematura destrucción de eritrocitos, que puede causar anemia.
- La cirrosis es un síntoma infrecuente.
- Síntomas neurológicos (que ocurren solo en algunas variantes de la enfermedad)
- Tipo II: convulsiones, hipertonía, retraso mental, apnea.
- Tipo III: sacudidas musculares (mioclono), demencia, apraxia ocular.
- Trombocitopenia; disminución de la cantidad de plaquetas circulantes en el torrente sanguíneo por debajo de los niveles normales.
- Osteoporosis: en el 75 % de casos se desarrollan defectos óseos visibles debidos a la acumulación de glucosilceramida. Suele describirse una deformación en el fémur distal en forma de matraz Erlenmeyer (necrosis aséptica en la articulación femoral).
- Los anteriores síntomas generan manifestaciones tales como: fatiga generalizada, falta de energía y ánimo, abdomen distendido a causa del agrandamiento de los órganos, dolor abdominal, retardo del crecimiento y desarrollo óseo. En la piel se puede observar una pigmentación café-amarilla y algunas manchas redondas y lisas.
Diagnóstico
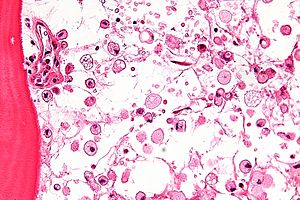
El diagnóstico de la GD se basa en la demostración de la deficiencia de actividad de la enzima glucosilceramidasa en leucocitos de la sangre periférica o de otras células nucleadas. Las pruebas de portador se realizan mediante un ensayo de actividad de la enzima, aunque no es fiable debido a la superposición de la actividad enzimática entre alelos portadores y no portadores de la mutación en el gen GBA. La identificación de los dos alelos que causan enfermedades en el gen GBA, el cual es el único gen en el que las mutaciones son conocidas por causar GD, proporciona una confirmación adicional del diagnóstico, pero no debe nunca sustituir a las pruebas bioquímicas de actividad enzimática.
Tratamiento
Para personas que no reciban la terapia de reemplazo enzimático (ERT) o la terapia de reducción de sustrato (SRT), el tratamiento sintomático incluye la esplenectomía parcial o total de esplenomegalia masiva y trombocitopenia. Todos los individuos afectados pueden y deben incluir a su tratamiento: la transfusión de sangre para anemia severa y hemorragias, la toma de analgésicos para el dolor de huesos, cirugía de reemplazo articular para el alivio del dolor crónico, en casos muy graves, y la toma de bifosfonatos orales y calcio para la osteopenia.
Prevención de las principales manifestaciones: la terapia de reemplazo enzimático es generalmente bien tolerada y proporciona la enzima exógena suficiente para superar el bloqueo en la vía catabólica. Personas con graves casos de GD, principalmente aquellos con afectación neurológica crónica (tipo 3), pueden beneficiarse de la médula ósea de un donante (BMT). Miglustat puede estar indicada en personas con síntomas de tipo 1 que no son capaces de recibir ERT.
Consejo genético
La enfermedad de Gaucher (EG) se hereda de forma autosómica recesiva. Si ambos progenitores presentan la mutación, los descendientes tiene una probabilidad del 25 % de estar afectados, un 50 % de probabilidad de ser portadores asintomáticos, y un 25 % de probabilidad de no estar afectados ni ser portadores. Se pueden realizar análisis específicos para investigar si existe la mutación y detectar a los portadores o enfermos en poblaciones de alto riesgo.
Las pruebas prenatales para embarazadas con riesgo son posibles mediante un ensayo de la actividad enzimática glucosylceramidase y las pruebas genéticas moleculares cuando ambas mutaciones que causan enfermedades en una familia son conocidos.
Otras lipidosis
- Enfermedad de Niemann-Pick.
- Enfermedad de Fabry.
- Enfermedad de Wolman.
- Xantomatosis cerebrotendinosa.
- Sitosterolemia.
- Enfermedad de Refsum.
- Enfermedad de Tay-Sachs.
- Leucodistrofia metacromática.
http://www.ncbi.nlm.nih.gov/books/NBK1269/
| Control de autoridades |
|
|---|
-
 Datos: Q861645
Datos: Q861645