
| Tumor de Wilms | ||
|---|---|---|
 Células tumorales (las células estrelladas) de un nefroblastoma. Microscopía electrónica.
| ||
| Especialidad | oncología | |
| Sinónimos | ||
| ||
Un nefroblastoma o tumor de Wilms es una neoplasia maligna del riñón y el segundo tipo más frecuente de cáncer abdominal en niños, después del neuroblastoma de glándula suprarrenal. Se presenta típicamente en la infancia (1 de cada 200 000 a 250 000 niños) y es muy infrecuente en mayores de 8 años así como en recién nacidos. Toma el nombre de Max Wilms (1867-1918), cirujano alemán que lo identificó por primera vez.
Un 75 % de los casos se presentan en niños sanos, mientras un 25 % se asocian a anormalidades del desarrollo como malformaciones en el tracto urinario, ausencia del iris (aniridia) y hemihipertrofia (crecimiento desproporcionado de un lado del cuerpo). Este tumor responde muy bien al tratamiento médico, citándose unas tasas de supervivencia del 90% al cabo de 5 años.
Fisiopatología
La mayoría de los nefroblastomas son unilaterales, siendo bilaterales en menos del 5 % de los casos. Se ubican con más frecuencia en el polo superior del riñón; tienden a ser tumores encapsulados y vascularizados que no rebasan la línea media hacia el lado opuesto del abdomen. Cuando existe metástasis tiende a ser en el pulmón.
Un tumor de Wilms puede desgarrarse, poniendo al paciente en un importante riesgo de hemorragia y diseminación por el peritoneo del tumor. De ocurrir un desgarro o rotura del tumor se debe expeditar la intervención quirúrgica por un cirujano con experiencia en la remoción de tumores de tal fragilidad.
Patogenia

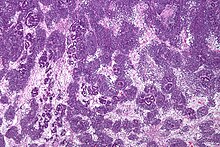
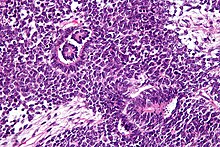
Patológicamente, un nefroblastoma consta de tres elementos:
- Blastema: una masa de células indiferenciadas capaces de crecimiento y regeneración en los órganos o partes del cuerpo, normalmente se encuentran en las primeras etapas de un organismo de desarrollo embrionario, y en la regeneración de tejidos, órganos y huesos.
- Mesénquima: un tipo de tejido conjuntivo reticular, que es de origen mesodérmico y situada en el mesodermo embrionario.
- Epitelio: el grupo de células que alinean las superficies del tumor.
El tumor de Wilms es un tumor maligno que contiene blastema metanéfrico, epitelio y estroma. Es característica su presencia en los glomérulos y túbulos renales rodeado por un estroma celular. El estroma puede incluir músculo estriado, cartílago, hueso, tejido graso y tejido fibroso. El tumor suele comprimir el parénquima renal normal.
El componente mesenquimal puede incluir células que muestran diferenciación rabdomioide, el cual puede por sí mismo mostrar elementos de malignidad: nefroblastoma rabdomiosarcomatoso.
Tumor de Wilms pueden ser separados en 2 grupos de pronósticos basados en las características patológicas:
- Favorable - Contiene componentes celulares bien desarrollados
- Anaplásicos - Contiene anaplasia difusa (escaso desarrollo de las células)
El tumor de Wilms ocurre con una frecuencia elevada en pacientes con aniridia, criptorquidia, hipospadias y otros trastornos congénitos genitourinarios. El riesgo de desarrollar este tumor es también aumentado en niños con el síndrome de Beckwith Wiedemann, el síndrome de Denys-Drash, el síndrome de Bloom y el síndrome WAGR.
Genética

Se ha notado una asociación genética en muchos casos de nefroblastoma. A comienzos de los años 1970 se propuso un modelo genético como parte de la etiología de este tipo de cáncer, fundamentando en un gen de supresión tumoral alterado denominado WT1 localizado en el brazo corto del cromosoma 11p13. Los cambios en este cromosoma predisponen al portador a un alto riesgo de trastornos congénitos, incluyendo el nefroblastoma. Otros candidatos involucrados en la patogenia de esta enfermedad neoplásica renal incluyen las proteínas IGFII, H19 y p57kip2 y se han implicado otros cromosomas como el 16, 7 y 17. Al menos la mitad de los pacientes con un tumor de Wilms y mutaciones en WT1 también cursan con mutaciones en el gen CTNNB1, que codifica el protooncogen β-catenina.
Un trabajo de investigación demostró que hasta un 30% de los pacientes estudiados con nefroblastoma tenían un gen inactivado en el cromosoma X, llamado WTX. La mayoría de los casos no cursan con mutaciones en ninguno de estos genes.
Cuadro clínico
En un 95 % de los casos el tumor se presenta como una masa palpable. La mayoría de los pacientes con un tumor de Wilms tienen poca afectación general. Algunos de los síntomas son inespecíficos:
- Dolor abdominal
- Masa abdominal
- Hematuria (presencia de sangre en la orina)—en menos del 10% de los casos
- Fiebre
- Anorexia (pérdida del apetito)
- Náuseas y/o vómitos
- Astenia
- Presión arterial elevada
- Estreñimiento
Diagnóstico
Cuando el examen físico de un niño revela la presencia de una masa abdominal, se indica un ultrasonido a ser completado en las siguientes 24 horas por un radiólogo con experiencia pediátrica. Si el ultrasonido no esclarece el origen de la lesión, se sugiere correlacionar con una tomografía. De confirmarse un tumor de origen renal, se debe de inmediato referir a un oncólogo pediatra con experiencia quirúrgica.
Tratamiento
Primero es obligatorio conocer la extensión de la enfermedad para optimizar el plan terapéutico. La extirpación quirúrgica del tumor es necesaria, así como otros tejidos adyacentes que puedan estar afectados. Desde la administración de radioterapia coadyuvante las sobrevida de pacientes con este tipo de cáncer mejoró en un 40 % y desde el comienzo del uso de quimioterapia se han visto tasas de sobrevivencia entre un 80 y 90 %.
Estadiaje
El estadiaje se determina por combinación de estudios de imagen, hallazgos patológicos y si el tumor es operable. El tratamiento se determina por las diferentes etapas del estadiaje:
Etapa I (43 % de los pacientes)
Durante la etapa I del tumor de Wilms, uno o más de los siguientes criterios deben cumplirse:
- El tumor está limitado a los riñones y puede ser resecado por completo
- La superficie de la cápsula renal está intacta
- El tumor no está roto ni ha sido biopsiado (abierta o por aguja) antes de su resección
- No hay asociación de los vasos renales
- No hay tumor residual evidente más allá de los márgenes de la escisión
Tratamiento: Nefrectomía + 18 semanas de quimioterapia
Pronóstico: el 98 % de los niños sobreviven al menos 4 años, sobrevida de 4 años es de 85% en caso de tumores anaplásicos
Etapa II (23 % de los pacientes)
En la Fase II del tumor de Wilms, uno o más de los siguientes criterios deben cumplirse:
- El tumor se extiende más allá del riñón, pero es extirpado por completo
- Ausencia de tumor residual aparente más allá de los márgenes de la escisión quirúrgica
- Cualquiera de las condiciones siguientes también pueden co-existir:
- Extensión tumoral fuera del parénquima renal y/o los vasos sanguíneos renales
- El tumor ha sido biopsiado antes de la remoción quirúrgica o hay derrame local del tumor durante la cirugía, confinado al flanco
Tratamiento: Nefrectomía + radiación abdominal + 24 semanas de quimoterapia
Pronóstico: supervivencia al cabo de 4 años 95%, sobrevida de 4 años es de 70% en caso de tumores anaplásicos
Etapa III (23 % de los pacientes)
En la Fase III de tumor de Wilms, uno o más de los siguientes criterios deben cumplirse:
- Tumor primario irresecable
- Metástasis ganglionar
- Márgenes quirúrgicos positivos
- Asociación de derrame tumoral en las superficies peritoneales, ya sea antes o durante la cirugía
Tratamiento: Radiación abdominal + 24 semanas de quimoterapia + nefrectomía después reducción tumoral
Pronóstico: supervivencia al cabo de cuatro años 95 %, sobrevida de cuatro años es de 70 % en caso de tumores anaplásicos...
Etapa IV (10 % de los pacientes)
La etapa IV del tumor de Wilms se define como la presencia de metástasis hematógena (pulmón, hígado, hueso, o el cerebro), o metástasis ganglionares abdomenopelvicos fuera de la región renal.
Tratamiento: Nefrectomía + radiación abdominal + 24 semanas de quimioterapia + radiación de zonas con metástasis de ser apropiado
Pronóstico: supervivencia al cabo de cuatro años 90 %, sobrevida de cuatro años es de 17 % en caso de tumores anaplásicos
Fase V (5 % de los pacientes)
Fase V tumor de Wilms se define como participación renal bilateral en el momento del diagnóstico inicial.
Nota: Para los pacientes con afectación bilateral, se debe llevar a cabo el estadiaje de cada uno de los lados de acuerdo con los criterios anteriores (etapa I a III) basados en la extensión de la enfermedad antes de la biopsia. La sobrevivencia al cabo de cuatro años es de 94 % para aquellos pacientes cuya lesión no es más avanzada que una fase I o fase II, 76 % para aquellos cuya lesión más avanzada era la etapa III.
Tratamiento: tratamiento individualizado basado en la carga tumoral
Fase I-IV anaplasia
Los niños con tumores anaplásicos fase I tienen un excelente pronóstico (sobrevida de 80-90 % al cabo de cinco años). Estos pueden ser abordados con el mismo régimen de la etapa I de pacientes con histología favorable.
Los niños con fase II a fase IV con anaplasia difusa, representan un grupo de alto riesgo. Estos tumores son más resistentes a la quimioterapia tradicional que los niños con tumor de Wilms con histología favorable, y requieren regímenes más agresivos.
Véase también
Enlaces externos
- National Wilms Tumor Study Group (en inglés)
| Control de autoridades |
|
|---|
-
 Datos: Q756289
Datos: Q756289
-
 Multimedia: Wilms' tumor / Q756289
Multimedia: Wilms' tumor / Q756289