
La síntesis de carbohidratos es un subcampo de la química orgánica que se ocupa específicamente de la generación de estructuras de carbohidratos naturales y no naturales. Esto puede incluir la síntesis de residuos de monosacáridos o estructuras que contienen más de un monosacárido, conocidos como oligosacáridos.
Antecedentes
En términos generales, los carbohidratos se pueden clasificar en dos grupos, azúcares simples y carbohidratos complejos. Los azúcares simples, también llamados monosacáridos, son carbohidratos que no se pueden convertir en azúcares más pequeños por hidrólisis. Cuando dos o más unidades de monosacáridos se conectan entre sí a través de un enlace glucósido, se forman carbohidratos complejos. Los carbohidratos complejos, de acuerdo con el número diferente de unidades de monosacáridos, se pueden clasificar en tres grupos, disacáridos, oligosacáridos y polisacáridos. Un disacárido se forma a partir de dos monosacáridos. Los oligosacáridos pueden estar formados por un pequeño número de monosacáridos unidos entre sí. Los oligosacáridos superiores se denominan polisacáridos. Ahora se sabe que los glicoconjugados juegan un papel indispensable en muchos procesos biológicos. Estos procesos biológicos en los que están involucrados los carbohidratos están típicamente asociados no a monosacáridos, sino a estructuras de oligosacáridos de glicoconjugados. Por lo tanto, la síntesis de oligosacáridos se vuelve cada vez más importante en el estudio de las actividades biológicas.
Síntesis de oligosacáridos
Los oligosacáridos tienen diversas estructuras. El número de monosacáridos, el tamaño del anillo, las diferentes estereoquímicas anoméricas y la existencia de azúcares de cadena ramificada contribuyen a la increíble complejidad de las estructuras de oligosacáridos. La esencia de la síntesis reductora de oligosacáridos es conectar el hidroxilo anomérico de los donantes de glicosilo a los grupos hidroxilo alcohólicos de los aceptores de glicosilo. La protección de los grupos hidroxilo del aceptor con el grupo hidroxilo alcohólico diana sin protección puede asegurar el control regioquímico. Además, factores como los diferentes grupos protectores, el disolvente y los métodos de glicosilación pueden influir en las configuraciones anoméricas. Este concepto se ilustra mediante una síntesis de oligosacáridos en el Esquema 1. La síntesis de oligosacáridos normalmente consta de cuatro partes: preparación de los donantes de glicosilo, preparación de los aceptores de glicosilo con un solo grupo hidroxilo desprotegido, el acoplamiento de ellos y el proceso de desprotección.

Bloques de construcción
Los donantes comunes en la síntesis de oligosacáridos son los haluros de glucosilo, los acetatos de glucosilo, los tioglicosidos, los tricloroacetimidatos, los glucósidos de pentenilo y los glucales. De todos estos donantes, los haluros de glucosilo son donantes clásicos, que desempeñaron un papel histórico en el desarrollo de reacciones de glucosilación. Los donantes de tioglicosido y tricloroacetimidato se usan más que otros en los métodos contemporáneos de glicosilación. Cuando se trata del método del tricloroacetimidato, una de las ventajas es que no es necesario introducir reactivos de metales pesados en el proceso de activación. Además, el uso de diferentes bases puede conducir selectivamente a diferentes configuraciones anoméricas. (Esquema 2) En cuanto a los tioglucósidos, la mayor fortaleza es que pueden ofrecer una protección temporal al centro anomérico porque pueden sobrevivir después de la mayoría de los procesos de activación. Además, se pueden emplear una variedad de métodos de activación, como NIS / AgOTf, NIS / TfOH, IDCP (perclorato de dicollidina de yodo), yodo y Ph2SO / Tf2O. Además, en la preparación del enlace glucosídico 1, 2-trans, el uso de tioglicosidos e imidatos puede promover la reorganización de los subproductos de ortoéster, ya que las mezclas de reacción son suficientemente ácidas.

Estereoselectividad
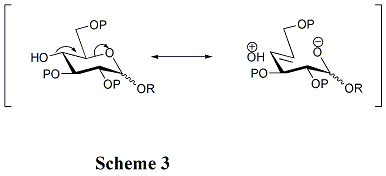
Las estructuras de los aceptores juegan un papel crítico en la tasa y estereoselectividad de las glicosilaciones. Generalmente, los grupos hidroxilo no protegidos son menos reactivos cuando están entre grupos protectores voluminosos. Esa es la razón por la cual el grupo hidroxilo en OH-4 en los piranosidos no es reactivo. La hiperconjugación está involucrada cuando OH-4 es anti-periplanar al oxígeno del anillo, lo que también puede reducir su reactividad. (Esquema 3) Además, los grupos protectores de acilo pueden reducir la reactividad de los aceptores en comparación con los grupos protectores de alquilo debido a su capacidad de extracción de electrones. El grupo hidroxilo en OH-4 de los derivados de N-acetilglucosamina es particularmente no reactivo.
El enlace glucosídico se forma a partir de un donante de glucosilo y un aceptor de glucosilo. Existen cuatro tipos de enlaces glucosídicos: 1, 2-trans-α, 1, 2-trans-beta, 1, 2-cis-α y 1, 2-cis-beta. Los enlaces glucosídicos 1, 2-trans se pueden lograr fácilmente utilizando donantes de glucosilo acilados con 2-O (participación del grupo vecino). Para evitar la acumulación de los intermedios de ortoéster, la condición de glicosilación debe ser ligeramente ácida.
Enlaces difíciles
Es algo más difícil preparar enlaces 1, 2-cis-β-glucosídicos estereoselectivamente. Típicamente, cuando los grupos no participantes en la posición O-2, el enlace 1, 2-cis-β se puede lograr mediante el uso de métodos de iones de haluro históricamente importantes, o mediante el uso de donantes de glicosilo alquilados con 2-O, comúnmente tioglicosidos o tricloroacetimidatos , en solventes no polares. A principios de la década de 1990, todavía era el caso de que el enlace beta-manósido era demasiado desafiante para que los aficionados lo intentaran. Sin embargo, el método introducido por Crich (Esquema 4), con protección de 4,6-bencilideno, un requisito previo y alfa triflato anomérico, un intermediario clave, deja este problema esencialmente resuelto. El enfoque de administración de aglicones intramoleculares (DAI) desarrollado de manera concurrente pero bastante más prolongado es una alternativa poco utilizada pero, sin embargo, estereoespecífica. is a little used but nevertheless stereospecific alternative.

Véase también
| Control de autoridades |
|
|---|
-
 Datos: Q5037882
Datos: Q5037882
-
 Multimedia: Carbohydrate synthesis / Q5037882
Multimedia: Carbohydrate synthesis / Q5037882