
| Síndrome de Walker-Warburg | ||
|---|---|---|
| ||
| Especialidad |
oftalmología neurología genética médica |
|
| Sinónimos | ||
|
Síndrome de Warburg; Síndrome de Chemke; Síndrome HARD; Disgenesia cerebroocular; Síndrome de distrofia muscular-displasia cerebroocular. | ||
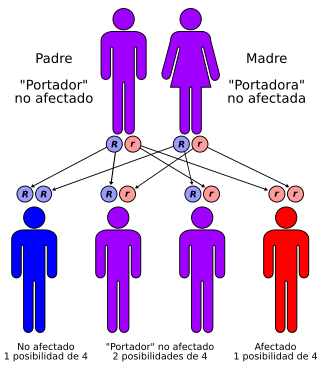
El síndrome de Walker-Warburg (WWS), también llamado síndrome de Warburg, síndrome de Chemke, síndrome HARD (hidrocefalia, agiria y displasia retiniana), síndrome de Pagon, disgenesia cerebroocular (DQO) o síndrome de distrofia muscular-displasia cerebroocular (COD-MD), es una forma rara de distrofia muscular congénita que sigue un patrón de herencia autosómica recesiva. Además se asocia con anomalías en el cerebro, cerebelo y ojos. Es la forma más grave de distrofia muscular congénita, muriendo la mayoría de los afectados antes de cumplir los tres años.
Epidemiología
Su distribución es mundial. Estudios en el Nordeste de Italia han informado de una tasa de incidencia de 1,2 por cada 100.000 nacidos vivos, aunque la prevalencia estimada es de 1'65 afectados por cada 100000 nacidos.
Etiopatogenia
El complejo distrofina glicoproteína (DGC) es un conjunto de proteínas que abarcan el sarcolema del músculo esquelético. Los defectos en el DGC parecen desempeñar un papel crítico en varias distrofias musculares debido a la interrupción de la organización de la membrana basal. El distroglicano es un propéptido expresado en muchos tipos celulares que sufre una glicosilación post-traduccional para formar, entre otros, α-distroglicano, la glicoproteína unida a la distrofina. Las mutaciones que producen un descenso en la glicosilación son las implicadas en el WWS. Estas mutaciones afectan a los genes POMT1 (9q34), POMT2 y el gen fukutina o FCMD (9q31). Las mutaciones de los genes implicados detectados no se hallan en todos los pacientes, por lo que existen además otros genes aún no identificados.
Cuadro clínico
Los síntomas y signos ya pueden estar presentes en el momento del nacimiento y primera infancia. Este síndrome incluye hipotonía generalizada, debilidad muscular, retraso en el desarrollo con retraso mental y, en algunos niños, convulsiones. Existen una amplia variedad de anomalías del ojo que pueden estar presentes, entre las que se incluyen cataratas, microcórnea y microftalmia, defectos del cristalino, desprendimiento de retina, coloboma y displasia, hipoplasia o atrofia del nervio óptico y la mácula. El glaucoma y la bulftalmia (aumento considerable del volumen ocular) pueden estar presentes. Las anomalías del encéfalo que pueden detectarse en los afectados son lisencefalia tipo II, hidrocefalia, hipoplasia vermal o general cerebelosa y tronco del encéfalo plano con pequeñas pirámides. La sustancia blanca muestra hipomielinización. Se han descrito en afectados otras anomalías adicionales del cerebro tales como hipoplasia o agenesia del cuerpo calloso, encefalocele occipital y la malformación de Dandy Walker. Algunas anomalías asociadas que afectan a otros sistemas son pene pequeño, testículos no descendidos, y, en raras ocasiones, otros rasgos faciales dismórficos tales como implantación baja de las orejas o que estén prominentes y labio leporino y/o paladar hendido.
Diagnóstico
Aparte de los signos clínicos, los hallazgos laboratoriales suelen presentar elevados niveles de creatina quinasa e hipoglicosilación de α-distroglicano. El diagnóstico molecular prenatal es posible en las familias con mutaciones conocidas. La ecografía prenatal puede ser útil para el diagnóstico en las familias donde el defecto molecular es desconocido, detectando hidrocefalia a las 13 semanas de gestación y encefalocele a las 18.
Tratamiento
No hay tratamiento específico disponible. Las convulsiones deben tratarse con anticonvulsivos, se pueden realizar procedimientos quirúrgicos para tratar la hidrocefalia y el encefalocele y terapia física para prevenir el empeoramiento de las contracturas musculares. En algunos casos, la alimentación debe ser controlada y suplementaria por sonda nasogástrica o gástrica.
Epónimo
WWS lleva el nombre de Arthur Earl Walker y Mette Warburg. Sus nombres alternativos incluyen el síndrome de Chemke y el síndrome de Pagon, llamada así por Juan M. Chemke y Roberta A. Pagon.
Enlaces externos
-
 Datos: Q1629483
Datos: Q1629483