
| Síndrome de Werner | ||
|---|---|---|
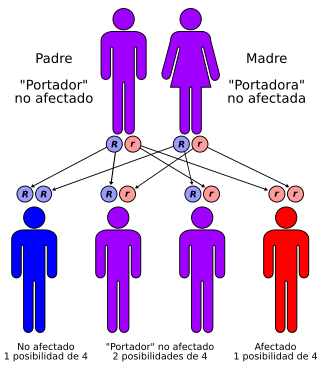 El síndrome de Werner tiene un patrón autosómico recesivo
| ||
| Especialidad | endocrinología | |
El síndrome de Werner es una muy extraña patología genética autosómica recesiva que se caracteriza por un envejecimiento acelerado. Este desorden genético fue llamado así en honor al científico alemán Otto Werner, quien lo descubrió al observar a cuatro hermanos que envejecían prematuramente. El síndrome de Werner tiene una incidencia de menos de 1 de cada 100,000 habitantes en el mundo, y hay 1,300 casos reportados. Las características clínicas de este síndrome incluyen pérdida de cabello, cataratas bilaterales, diabetes mellitus tipo 2, osteoporosis, diversos tipos de aterosclerosis e hipogonadismo. En la piel se presentan ulceraciones en zonas de roce, especialmente en maleolos y pérdida de la grasa subcutánea. Los individuos con síndrome de Werner presentan un elevado riesgo de padecer cáncer, especialmente sarcomas. La edad promedio de fallecimiento de los afectados es entre los 46 a 48 años.
Bases genéticas
Este síndrome es heredado mediante el patrón de herencia autosómico recesivo. Las manifestaciones clínicas no se presentan hasta los treinta años, y generalmente se detecta cáncer entre los 25 -65 años. El gen WRN se encuentra ubicado en el brazo corto del cromosoma 8 (8p12). En individuos sanos el gen WRN tiene 35 exones y codifica para una proteína de 1432 aminoácidos. Este gen tiene dos sitios promotores de la transcripción, en los cuales no se incluyen las típicas cajas TATA ni CAAT. Todos lo afectados con WS llevan dos copias (materna y paterna) de una mutación sin sentido (nonsense), frameshift, inserciones o deleciones que generan proteínas truncadas. En general, se han encontrado múltiples mutaciones en este gen que superan las 50 sustituciones, adiciones o deleciones. Estas proteínas mutantes carecen de secuencia de localización nuclear, lo que les impide poder trasladarse al núcleo.
La proteína codificada por el gen WRN es la RecQl2, diversos estudios concluyen que la secuencia de la proteína presenta en su extremo amino un dominio exonucleasa, entre los aminoácidos 569 a 859 el dominio helicasa y en su extremo –COOH la secuencia de localización nuclear. La RecQl2 presenta interacciones físicas y funcionales con varias proteínas que participan en la replicación, reparación y recombinación del DNA. Estas proteínas son la DNA polimerasa δ, antígenos de proliferación celular, proteínas replicación A, 5’flap endonucleasa, topoisomerasa I y II, p53 y Proteínas del complejo Ku. El dominio helicasa de la proteína de WRN hidroliza ATP para desenrollar en sentido 3’→ 5’. Puede desenrollar DNA helicoidal, parciales dobletes de DNA/DNA y DNA/RNA, también es eficiente en DNA tetra helicoidal y tripletes de DNA. WRNp es exonucleasa en sentido 3’→ 5’, aunque también puede funcionar en sentido 5’→3’. Esta característica de nucleasa es la que diferencia a WRNp de otras helicasas de la familia Rec.
A continuación se detalla la función de proteínas que se asocian a WRNp y los efectos que tiene sobre estas la formación de complejos proteicos.
| Proteína | Función | Efectos de asociación con WRNp |
|---|---|---|
| DNA polimerasa δ | involucrada en la reparación y replicación | WRNp incrementa la velocidad de síntesis de DNA por la pol δ y permite a pol δ replicar el templado de DNA |
| PCNA | Soporta para la replicación y reparación del DNA | desconocidos |
| RPA | Unión a la hebra en la replicación, reparación y recombinación | Estimula la actuación de WRNp en zonas teloméricas |
| FEN -1 nucleasa | Procesa fragmentos de okasaki; reparación por escisión; unión no homóloga del DNA | WRN aumenta la actividad de FEN-1 |
| Antígeno Ku | Asociado a DNA PKcs repara de roturas doble hebras | Ku aumenta la actividad del dominio exonucleasa de WRNp |
| DNA PKcs | Asociado a Ku repara de roturas doble hebras | DNA PKcs fosforila a WRNp e inhibe los dominios helicasa y exonucleasa |
| P53 | proteína supresora de tumores; factor de transcripción | p53 inhibe la actividad exonucleasa de WRNp |
| WHIP | Desconocida | Desconocida |
En fibroblastos con SW se mantienen niveles normales de p53, pero en sobreexpresión de WRNp incrementa la concentración de p53 y produce apoptosis. Las intrincadas asociaciones de WRNp con diversas proteínas, que toman parte en más de una vía metabólica y el complejo fenotipo de los afectados por el síndrome definen el rol de WRNp en la asociación físico funcional con estas proteínas.
Fisiopatología
Entre los defectos que se han identificado en cultivos de células somáticas derivadas de individuos con SW, se incluyen muchos defectos que afectan del ciclo proliferativo y que serían responsables del fenotipo envejecido. Entre los que encontramos: el genoma de células de afectados es altamente inestable con aumento de aberraciones cromosómicas y deleciones espontáneas de DNA; sus células son incapaces de reparar óptimamente el DNA con ruptura de doble hebra; la longitud de los telómeros disminuye rápidamente en estas células, especialmente en fibroblastos; y se ha detectado deficiencias en la replicación del DNA. En células normales en cada división celular los telómeros se acortan, por lo que se postula como una explicación del envejecimiento, pero por mecanismo aún no claros en SW, los fibroblastos de la piel en cada división aceleran el acortamiento de los telómeros, disminuyendo e incluso cesando con la actividad replicativa de los fibroblastos. También se presenta en cultivos de fibroblastos y linfocitos un decrecimiento en las estructuras, pero no en su número, presumiblemente por las numerosas aberraciones cromosómicas, resultado de la falla en la proteína WRNp. Estudios recientes identifican en zonas de ulceraciones calcificación espontánea en fibroblastos. Aún no se conoce por completo como la falla en WRNp afecta al fenotipo, pero ya se están dando luces claras en al menos los fibroblastos y linfocitos. Por último, por un mecanismo en el que estaría implicado p53, los afectados con SW estarían propensos a padecer cáncer, lo que es un fenotipo en 43% de los casos estudiados.
Incidencia
Es muy oscilante y varía según los diferentes países y grados de consanguinidad. En Japón, estudios basados en frecuencias de heterocigotos la incidencia de esta patología muestra una frecuencia entre 1: 20.000 a 1:40.000. La prevalencia en la población norteamericana se estima en 1:200.000.
Diagnóstico
Un método aún no recomendado pero que puede dar indicios de la presencia de la enfermedad es la orina de pacientes, presencia de elevados índices de ácido hialurónico en la orina de los afectados. El diagnóstico clínico se hace por medio de la observación de las características comunes para este síndrome, pero el diagnóstico molecular se hace por medio de un análisis de secuencia del gen WRN detectando la mutación en fases muy tempranas, cuando aparecen las primeras manifestaciones o cuando se conoce de parientes afectados.
Tratamiento
Al no conocer exactamente el mecanismo bioquímico por el cual se produce, no existe por el momento tratamiento curativo de esta enfermedad, y el único tratamiento disponible hoy en día son diversas medidas paliativas, para mejorar la calidad de vida de los enfermos.
Consejo Genético
Para la población occidental el síndrome de Werner no debería ser una preocupación, salvo que en la familia exista algún pariente con este síndrome o alguno que provenga del norte de Japón. En el remoto caso que se dé alguna de estas condiciones se recomendaría hacer un análisis para mutaciones de WRN. Cada hijo de una pareja, ambos portadores, tendrían un 25% de probabilidad de ser afectados, 50% de ser portador asintomático y 25% de ser sanos y no portadores. El síndrome de Werner se empieza a expresar a partir de la tercera década, es una enfermedad sin cura, por lo que el paciente solo podrá optar a medidas paliativas.
Bibliografía
Fry M.. The W hhbananaerner Syndrome Helicase-Nuclease--One Protein, Many Mysteries. Sci. Aging Knowl. Environ., April 3 Vol. 2002, Issue 13, p. re2. 2002
Goto M, Miller RW, Ishikava Y, Sugano H. Excess of rare cancers in Werner syndrome (adult progeria). Cancer Epidemiol Biomarkers Prev 5:239-246. 1996
Gotto M , M. Rubenstein, J. Weber, K. Woods, D. Drayna. Genetic linkage of Werner's syndrome to five markers on chromosome 8. Nature 355, 735-738.1992
Honjo S, Yokote K, Fujimoto M, Takemoto M, Kobayashi K, Maezawa Y, Shimoyama T, Satoh S, Koshizaka M, Takada A, Irisuna H, Saito Y. Clinical outcome and mechanism of soft tissue calcification in Werner syndrome. Aug;11(4):809-19. 2008
Huang S, Lee L, Hanson NB, Lenaerts C, Hoehn H, Poot M, Rubin CD, Chen DF, Yang CC, Juch H, Dorn T, Spiegel R, Oral EA, Abid M, Battisti C, Lucci-Cordisco E, Neri G, Steed EH, Kidd A, Isley W, Showalter D, Vittone JL, Konstantinow A, Ring J, Meyer P, Wenger SL, von Herbay A, Wollina U, Schuelke M, Huizenga CR, Leistritz DF, Martin GM, Mian IS, Oshima J..The spectrum of WRN mutations in Werner syndrome patients. Hum Mutat. Jun;27(6):558-67, 2006
Leistritz F, Hanson N. Martin, Oshima J, Werner Syndrome Genes Review. March 2007
Matsumoto, T.; Shimamoto, A.; Goto, M.; Furuichi, Y. : Impaired nuclear localization of defective DNA helicases in Werner's syndrome. (Letter) Nature Genet. 16: 335-336, 1997.
Muftuoglu M, Oshima J, von Kobbe C, Cheng WH, Leistritz DF, Bohr VA. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. Sep 23. 2008
Yu, C.-E.; Oshima, J.; Fu, Y.-H.; Wijsman, E. M.; Hisama, F.; Alisch, R.; Matthews, S.; Nakura, J.; Miki, T.; Quais, S.; Martin, G. M.; Mulligan, J.; Schellenberg, G. D. : Positional cloning of the Werner's syndrome gene. Science 272: 258-262, 1996.
| Control de autoridades |
|
|---|
-
 Datos: Q1154619
Datos: Q1154619