
| Síndrome de Klinefelter | ||
|---|---|---|
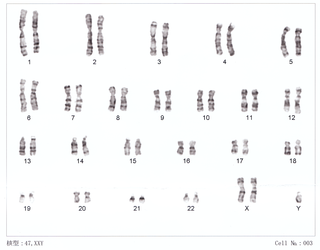 47,XXY
| ||
| Especialidad | genética médica | |
El síndrome de Klinefelter (SK) o 47,XXY es la caracterización clínica de una mutación cromosómica que afecta a hombres y que incluye, entre otras manifestaciones, hipogonadismo hipergonadotrópico, ginecomastia, dificultades en el aprendizaje e infertilidad. Se origina por la existencia de dos cromosomas X y un cromosoma Y. Es la enfermedad genética más común en varones. Algunos hombres no presentan síntomas y no saben que padecen esta condición hasta la edad adulta al presentarse infertilidad.
Se basa en una alteración genética que se desarrolla por la separación incorrecta de los cromosomas homólogos durante las meiosis que dan lugar a los gametos de uno de los progenitores, aunque también puede darse en las primeras divisiones del cigoto. Constituye la causa más frecuente de hipogonadismo masculino de carácter permanente.
El sexo está determinado por los cromosomas X e Y. Los machos tienen los cromosomas sexuales XY (46,XY) y las hembras, los cromosomas sexuales XX (46,XX). En el síndrome de Klinefelter, el macho cuenta, como mínimo, con un cromosoma X extra, dando lugar en el 75 % de los casos a un cariotipo (47,XXY). No obstante, aproximadamente un 20 % de los casos son mosaicos cromosómicos, con variantes como (48,XXXY), y (49,XXXXY) en el 5 % de los casos.
Se cree que Carlos II de España sufrió este síndrome, debido fundamentalmente a los sucesivos matrimonios endogámicos de sus antepasados.
Historia
El primer diagnóstico del síndrome no está del todo claro. Una teoría postula que los primeros casos se reportaron en el Egipto predinástico, ya que algunos huesos encontrados presentan algunos de los síntomas de la enfermedad; pero debido a que es fácil confundir otras enfermedades, este hecho no se toma como definitivo.
Fue descrito en 1942 por Harry Klinefelter y colaboradores, que estudiaron nueve pacientes con ginecomastia, testículos pequeños, azoospermia y elevada concentración de gonadotropinas. Estos estudiosos sugirieron que el defecto primario estaba en las células de Sertoli y propusieron que, además, en estos pacientes había una deficiencia en una hormona testicular que regulaba la concentración de gonadotropinas hipofisiarias, a la que llamaron "hormona X" o inhibina.
En 1956 se demostró la presencia del corpúsculo de Barr en pacientes con síndrome de Klinefelter y tres años más tarde se identificó que el cariotipo de un sujeto con la enfermedad era (47,XXY). De esta manera se estableció que la presencia de un cromosoma X extra era el factor etiológico fundamental para desarrollar las características de dicho síndrome (véase apartado Manifestaciones).
Etiología
El síndrome de Klinefelter también es conocido como 47- XXY y se considera la anomalía cromosómica más común en los humanos, presentándose con una incidencia de 1 en 1000 en los varones recién nacidos vivos. Los afectados presentan un cromosoma X supernumerario, lo que conduce a un fallo testicular primario cuyas consecuencias primarias son infertilidad e hipoandrogenismo. El síndrome de Klinefelter es considerado la causa más frecuente en hombres de hipogonadismo. El cromosoma X adicional en los pacientes con síndrome de Klinefelter a menudo es adquirido por un error en la disyunción durante la meiosis I (gametogénesis). El error en el proceso de separación de cromosomas durante la división celular se da cuando cromosomas homólogos (en este caso, los cromosomas sexuales X e Y) fallan al separarse, originando gametos (masculinos o femeninos) con 24 cromosomas, debido a dicho cromosoma adicional.

El exceso de cromosoma X proviene de la madre en aproximadamente la mitad de los casos y del padre en la otra mitad. La edad materna es el único factor de riesgo para el síndrome de Klinefelter: A partir de una edad materna de 40 años, el riesgo de tener un hijo con síndrome de Klinefelter es cuatro veces mayor que en las mujeres menores de 24 años.
La anomalía cromosómica puede originarse también por un error durante las divisiones mitóticas del cigoto, produciendo así los casos de mosaicismo.
En mamíferos con más de un cromosoma X (en el caso de humanos, mujeres), se da la inactivación de uno de los dos cromosomas, de modo que se equipare la carga génica con el hombre. Esto también ocurre en los varones XXY, aunque hay cierta evidencia que sugiere que algunos genes localizados en las regiones pseudoautosomales de sus cromosomas X presentan correspondencia con su cromosoma Y, siendo capaces de expresarse.
Los estudios en sujetos prepuberales (47,XXY) no muestran deficiencias en las concentraciones de las hormonas LH, FSH o testosterona, comparados con sujetos prepúberes (46,XY) y la respuesta a la gonadoliberina (LHRH, hormona hipotalámica liberadora de gonadotropinas) es normal en ambos grupos. Sin embargo, entre los 12 y 14 años de edad en los sujetos (47,XXY) las concentraciones de gonadotropinas se incrementan y la testosterona permanece en límites inferiores para la edad.
Patogenia
En biopsias realizadas a niños con el síndrome, se ha observado solo disminución en el número de células germinales. No obstante, después de la pubertad se aprecia hialinización y fibrosis de los túbulos seminíferos, que son los cambios histológicos característicos del síndrome y que originan disminución en el volumen testicular y aumento de su consistencia. Además, se observa ausencia de células germinales, hiperplasia y agregación de las células de Leydig, como respuesta a la hiperestimulación por la hormona LH. Las alteraciones histológicas se hacen más frecuentes con la edad.
La pérdida de túbulos seminíferos y células de Sertoli produce una disminución en las cifras de inhibina B, el factor regulador de FSH, y de AMH u hormona antimülleriana, lo que disminuye la retroalimentación negativa sobre la FSH, aumentando ésta. La ausencia de espermatogénesis es secundaria a la presencia de cromosomas supernumerarios, que se mantienen activos durante la gametogénesis.
A pesar de la generalidad extendida de que los varones que presentan síndrome Klinefelter van a desarrollar un determinado fenotipo, muchos de ellos no lo hacen, pudiendo llevar una vida normal. En estos casos, el síndrome se hará evidente en la edad adulta, cuando el individuo acuda al especialista por problemas de fertilidad, siendo entonces cuando se detecta el cromosoma extra y se diagnostica la causa de la esterilidad. Debido a estos casos, muchos médicos e investigadores están empezando a dejar en desuso el término "síndrome de Klinefelter", usando en su lugar la descripción de "varones XXY".
Cuadro clínico

El cuadro clínico del síndrome de Klinefelter se presenta en diversas manifestaciones y complicaciones, varía en ocasiones de un paciente a otro.
A continuación, se listan las características más comunes en los varones XXY. No obstante, no todas ellas aparecen en un mismo individuo:
- Bebés con síndrome de Klinefelter suelen gatear y comenzar a andar de forma más torpe y tardía que los demás niños.
- Retraso en el área del lenguaje, lectura y comprensión. Los niños XXY por lo general aprenden a hablar más tarde que los otros niños, y pueden tener ciertas dificultades para leer y escribir. Muchos de ellos suelen tener algún grado de dificultad con el lenguaje de por vida. Sin embargo, los varones XXY presentan un cociente intelectual normal.
- Talla elevada en la edad adulta.
- Mayor propensión a padecer cáncer de pecho, alteraciones venarias y osteoporosis.
- Tendencia al sobrepeso.
- En ocasiones criptorquidia o micropene.
- Casi siempre existe escroto hipoplásico.
- Esterilidad por azoospermia.
- Ginecomastia unilateral o bilateral. Se caracteriza por el desarrollo de pechos en el hombre (tejido mamario agrandado).
- Escasez de vello en la cara y en todo el cuerpo, consecuencia directa de la baja concentración de testosterona.
- Vello pubiano disminuido, o siguiendo un claro patrón femenino.
- Gonadotrofinas elevadas en la pubertad.
- Disminución de la libido sexual en la edad adulta
- Trastornos emocionales, ansiedad, depresión, etc.
- Falta de autoestima, debida en la mayoría de los casos a los caracteres femeninos perceptibles por el varón (ginecomastia, etc.).
Enfermedades concomitantes
Las personas con síndrome de Klinefelter tienen un mayor riesgo de diabetes mellitus tipo 2, osteoporosis, ginecomastia, la aparición de trombosis y cáncer de mama en comparación con la población general. Sin embargo, el riesgo de cáncer de seno está por debajo del riesgo normal para las mujeres. Además, los estudios indican un mayor riesgo de desarrollar depresión.
Diagnóstico
Las características físicas de un síndrome de Klinefelter pueden ser una estatura alta, vello corporal bajo y ocasionalmente un agrandamiento de la glándula mamaria. Por lo general, hay un pequeño volumen testicular de 1 a 5 ml por testículo (valores normales: 12 a 30 ml) e infertilidad. Durante la pubertad y la edad adulta, los niveles bajos de testosterona con niveles elevados de las hormonas pituitarias FSH y LH en la sangre pueden indicar la presencia del síndrome de Klinefelter. Un espermiograma también puede ser parte de la investigación adicional. A menudo hay una azoospermia, rara vez una oligospermia.Sin embargo, el síndrome de Klinefelter solo puede diagnosticarse de manera confiable mediante un análisis cromosómico al detectar un cromosoma X adicional. Una pequeña muestra de sangre es suficiente como material de prueba. Para determinar si tiene forma de mosaico, también se puede realizar un análisis cromosómico en las células de la mucosa oral.
Otra forma de diagnosticar un Klinefelter de forma prenatal es por amniocentesis o por la muestra del villus coriónico (CVS).
Tratamiento
La terapia causal no es posible porque el síndrome de Klinefelter es causado por una alteración cromosómica. Desde el inicio de la pubertad, la deficiencia de testosterona existente puede ser compensada por la terapia de sustitución hormonal adecuada. Preparaciones de testosterona están disponibles en forma de jeringas, parches o gel. Además, los hombres con síndrome de Klinefelter a menudo tienen una deficiencia de vitamina D. Por lo tanto, los niveles de vitamina D en adolescentes y adultos deben controlarse regularmente. Una sustitución de vitamina D conduce a una mejor densidad ósea. Si existe ginecomastia, existe la posibilidad de una mastectomía (extirpación quirúrgica del tejido mamario). Si niños con síndrome de Klinefelter tienen dislexia, dificultad para concentrarse o conducción débil, existe la posibilidad de medidas de apoyo como la terapia del hablar o la terapia ocupacional.
Oportunidades para la infertilidad
Los nuevos métodos de medicina reproductiva, específicamente la llamada inyección intracitoplasmática de espermatozoides (ICSI) con extracción previa de espermatozoides testiculares (TESE), han dado lugar a descendencia de varones con síndrome de Klinefelter. Un estudio pequeño encontró que los niños biológicos de los hombres con síndrome de Klinefelter (esperma obtenido por TESE) no tienen un cromosoma X adicional y, por lo tanto, no tienen síndrome de Klinefelter. Si no se pueden obtener espermatozoides funcionales a partir del semen o TESE, no hay posibilidad de producir hijos biológicos. Puesto que el tratamiento de testosterona suprimiría la espermatogénesis, la posibilidad de TESE con el paciente y sus padres debe ser discutido en consulta con los médicos asistentes antes del inicio de la terapia de testosterona. Si la terapia de testosterona ya se ha iniciado, pero el paciente se desea todavía una TESE, la terapia de testosterona debe pausarse durante al menos 6 meses para que la espermatogénesis puede recuperarse.
Directrices
En septiembre de 2020, la Academia Europea de Andrología (European Academy of Andrology) publicó por primera vez directrices para el síndrome de Klinefelter.
Véase también
| Control de autoridades |
|
|---|
-
 Datos: Q207133
Datos: Q207133
-
 Multimedia: Klinefelter's syndrome / Q207133
Multimedia: Klinefelter's syndrome / Q207133