
| Síndrome antifosfolípidos | ||
|---|---|---|
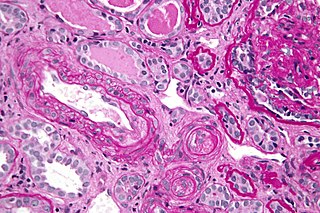
Micrografía mostrando una microangiopatía trombótica, tal como puede ser vista en un SAFL. Biopsia de riñón. Coloración PAS.
| ||
| Especialidad | hematología | |
El síndrome antifosfolípido o síndrome de anticuerpos antifosfolípidos (SAFL), también llamado a veces síndrome de Hughes, es un cuadro autoinmunitario de hipercoagulabilidad causado por anticuerpos dirigidos contra proteínas de unión a los fosfolípidos de las membranas celulares. Este cuadro se caracteriza por una mayor susceptibilidad a la formación de coágulos intravasculares (trombosis), tanto en arterias como en venas, y también por complicaciones obstétricas tales como abortos espontáneos, muerte fetal, partos pretérmino o preeclampsia severa.
El síndrome se debe a un trastorno autoinmunitario que lleva a la producción de autoanticuerpos dirigidos contra unos componentes de las membranas celulares llamados fosfolípidos (anticuerpos antifosfolípidos o aPL). En concreto, el cuadro se caracteriza por la aparición dos grupos de anticuerpos: los anticuerpos anticardiolipinas, dirigidos contra un componente de las membranas de las mitocondrias, la cardiolipina, y los denominados anticoagulante lúpico, un grupo heterogéneo de anticuerpos dirigidos contra complejos fosfolípido-proteína. Estos últimos han demostrado una alteración en la cascada de coagulación en ensayos in vitro, y entre ellos destacan los anticuerpos anti apolipoproteína H, también llamados anti-β2 glicoproteína I. El término "síndrome antifosfolípido primario" se utiliza cuando el SAFL aparece en ausencia de otras enfermedades autoinmunitarias, mientras que el término "síndrome antifosfolípido secundario" se utiliza cuando el SAFL aparece en el contexto de otras enfermedades autoinmunitarias tales como el lupus eritematoso sistémico (LES). En algunos casos raros, un SAFL puede conducir a un fallo multisistémico súbito debido a una trombosis generalizada; en este caso se suele utilizar el término síndrome antifosfolípido catastrófico, y conlleva un alto riesgo para la vida.
El síndrome antifosfolípidos se diagnostica por medio de análisis sanguíneos. Muy frecuentemente requiere tratamiento con anticoagulantes tales como la heparina o inhibidores de la vitamina K tales como la warfarina (comercializada con el nombre de Coumadin). La warfarina no se utiliza durante el embarazo, ya que es capaz de atravesar la placenta y presenta actividad teratogénica, por lo que en tales casos se utiliza la heparina.
Mecanismo
El síndrome antifosfolípido es una enfermedad autoinmunitaria en la cual los anticuerpos antifosfolípidos (anticuerpos anticardiolipinas y anticoagulante lúpico) reaccionan contra las proteínas que se unen a los fosfolípidos aniónicos de las membranas celulares. Como en muchas enfermedades autoinmunitarias, es más frecuente en las mujeres que en los hombres. Todavía no se conoce con certeza la etiología exacta, pero la activación del sistema de coagulación es un hecho evidente. Los anticuerpos antifosfolípidos de importancia clínica (aquellos cuyo aumento redunda en un proceso autoinmunitario) se encuentran asociados con trombosis y enfermedades vasculares.
Los anticuerpos Anti-β2-glicoproteína I (Anti-ApoH) son un subgrupo de anticuerpos lúpicos que se unen a la ApoH, lo que a su vez conduce a la inhibición de la proteína C, una glucoproteína que desempeña un papel regulatorio en la vía común de coagulación (lo hace degradando al factor V).
Los anticuerpos del anticoagulante lúpico se unen a la protrombina, conduciendo a un aumento en su clivaje hacia trombina, la forma activa.
En el síndrome antifosfolípido aparecen, además, anticuerpos dirigidos contra:
La proteína S, la cual es un cofactor de la proteína C, por lo que los anticuerpos anti proteína S disminuyen la eficiencia de la proteína C;
La anexina A5, la cual forma una especie de escudo en torno a las moléculas de fosfolípidos con carga negativa, reduciendo por lo tanto su capacidad de desencadenar una cascada de coagulación. Por lo tanto, los anticuerpos anti anexina A5 incrementan los pasos de la coagulación que son dependientes de fosfolípidos.
Los anticuerpos del anticoagulante lúpico son aquellos que presentan una asociación más estrecha con la trombosis, y entre ellos los que tienen como objetivo a la β2glicoproteína 1 presentan una mayor asociación que aquellos que tienen como objetivo a la protrombina. Los anticuerpos anticardiolipinas presentan asociación con las trombosis a títulos entre moderados y altos (>40 GPLU o MPLU). Los pacientes que presentan ambos tipos de anticuerpos lúpicos y títulos entre moderados y altos de anticuerpos anticardiolipinas son los que tienen el mayor riesgo de trombosis.
Signos y síntomas
La presencia de anticuerpos antifosfolípidos (aPL por sus siglas en inglés) en ausencia de trombosis o complicaciones en el embarazo no indica SAFL.
El síndrome antifosfolípido puede causar trombosis arteriales o venosas en cualquier sistema de órganos, o complicaciones en el embarazo. En pacientes con SAFL el trastorno venoso más frecuente es la trombosis venosa profunda de extremidades inferiores, mientras que el trastorno arterial más frecuente es el accidente cerebrovascular. En mujeres embarazadas afectadas por SAFL pueden ocurrir abortos espontáneos antes de las 20 semanas de gestación, mientras que después de 20 semanas predomina la preeclampsia. También pueden producirse infartos de placenta, partos prematuros y muerte fetal en mujeres con SAFL. En algunos casos, el síndrome antifosfolípido podría haber sido causa de retraso en el desarrollo o incluso de retraso mental en el recién nacido debido a una inhibición de los trofoblastos inducida por los anticuerpos antifosfolípidos. El síndrome antifosfolípido responsable de la mayor parte de los abortos prematuros en los trimestres finales es el asociado a lupus eritematoso sistémico.
Otros hallazgos frecuentes, a pesar de no ser parte del criterio de clasificación del SAFL, son: trombocitopenia, enfermedad de las válvulas cardíacas y livedo reticularis (un cuadro cutáneo). Algunos pacientes presentan cefaleas, migrañas y oscilopsia. Muchos pacientes con síndrome antifosfolípido primario tienden a desarrollar lupus eritematoso sistémico con el transcurso del tiempo.
Factores de riesgo
Los factores de riesgo para el desarrollo de un síndrome antifosfolípido incluyen:
- SAFL primario
- SAFL secundario
- LES u otro trastorno autoinmunitario
- marcadores genéticos: HLA-B8, HLA-DR2, HLA-DR3
Diagnóstico
Se debe sospechar un síndrome antifosfolípido en caso de un cuadro clínico tal como una trombosis venosa o arterial sin causa aparente, trombocitopenia y en caso de pérdidas fetales recurrentes. El diagnóstico definitivo se hace por laboratorio.
Las pruebas de laboratorio para el síndrome antifosfolípido se hacen sobre la base de dos ensayos, el ensayo para demostrar la presencia de anticoagulante lúpico y el ensayo de anticuerpos anticardiolipinas que se hace por ELISA
La trombofilia de origen genético es parte del diagnóstico diferencial en caso de SAFL, y puede coexistir en algunos pacientes con SAFL. La presencia de una trombofilia genética puede determinar la necesidad de terapia anticoagulante. Un panel para diferenciar trombofilia genética podría consistir en:
- Estudios para determinar la variante Factor V Leiden y mutaciones de la protrombina, niveles de factor VIII, mutaciones de la MTHFR (hiperhomocisteinemia).
- Niveles de proteína C, proteína S libre y total, antitrombina, plasminógeno, activador tisular del plasminógeno, y activador inhibidor del plasminógeno-1 (PAI-1)
La búsqueda de anticuerpos dirigidos contra cada uno de los posibles objetivos de los anticuerpos antifosfolípidos (por ejemplo anticuerpos anti-β2 glicoproteína 1 y anti fosfatidilserina) se encuentra actualmente en debate, ya que de momento el ensayo para anticuerpos anticardiolipinas parece ser más sensible y específico para el diagnóstico de SAFL, incluso a pesar de que las cardiolipinas no son consideradas un objetivo de los anticuerpos antifosfolipidos in vivo.
Anticoagulante lúpico
Estos anticuerpos se prueban utilizando un mínimo de dos ensayos de coagulación que sean sensibles a los fosfolípidos, esto debido a la naturaleza heterogénea de los anticuerpos del anticoagulante lúpico.
La realización de la prueba incluye un como mínimo un ensayo de cribado o screening y uno de confirmación. Pudiendo añadir además un ensayo para determinar inhibidores de los factores de coagulación.
Entre los primeros se pueden contar por ejemplo:
- aPTT
- Tiempo de coagulación con caolín
- Tiempo del veneno de víbora de Russel
- Si el test de screening da alterado se procede al siguiente paso.
Identificación del inhibidor:
Se considera la presencia de un inhibidor cuando no se observa corrección en el test de screening realizado con las mezclas de plasma normal (ver más abajo).
Test de confirmación: Para diferenciar al AL de los inhibidores específicos de factor, se utilizan ensayos basados en tres características diferentes:
- Concentración reducida de fosfolípidos para acentuar el efecto del inhibidor
- Concentración alta de fosfolípidos para neutralizar al inhibidor
- Configuración alterada de fosfolípidos para neutralizar al inhibidor
Un paneo general de laboratorio en un paciente se iniciaría por ejemplo haciendo un ensayo de APTT con un resultado prolongado que no se corrige con una mezcla al 80:20 con plasma humano normal en caso de ser positivo (las pruebas con mezclas al 50:50 son prácticamente insensibles a menos que existan niveles sumamente elevados de anticuerpos). Otras de las principales son el ensayo del veneno de la víbora de Rusell diluido (DRVVT), el tiempo parcial de tromboplastina con caolín (KPTT), tiempo de tromboplastina diluida (TDT/DTT), o tiempo de protrombina utilizando tromboplastina sensible al anticoagulante lúpico. Estas pruebas deben ser llevadas a cabo un mínimo de dos veces con al menos 6 semanas de mediación entre una y otra y demostrando una positividad persistente en ambas ocasiones para permitir el diagnóstico de síndrome antifosfolípido. Esto es para prevenir que pacientes con resultados positivos pero transientes (causados por ejemplo por una infección) sean diagnosticados como positivos.
En caso de que el ensayo de cribado de un resultado alterado se procede en segundo término distinguír si se trata del anticoagulante lúpico o un inhibidor específico de alguno de los factores de coagulación (p. ej. anti factor VII). Esto normalmente se consigue diferenciando los efectos de un anticuerpo específico. El anticoagulante lúpico, al actuar sobre los fosfolípidos, inhibe a todos los factores de la vía de activación por contacto (Factor VIII, Factor IX, Factor XI y Factor XII). El anticoagulante lúpico muy raramente causaría que un ensayo de factor de coagulación diera como resultado un valor menor a 35 UI/dL (35%), mientras que un antcuerpo específico raramente permitiría un resultado mayor a 10 UI/dL (10%).
Sin embargo, debido a los efectos anticoagulantes de los anticuerpos lúpicos, no se monitorea la terapia anticoagulante por medio del APTT, y esta situación es generalmente mejor desarrollada utilizando un ensayo cromogénico basado en la inhibición del Factor Xa por la antitrombina en presencia de heparina.
Anticuerpos anticardiolipinas
Estos anticuerpos pueden ser detectados utilizando un inmunoensayo de tipo ELISA, que busca la presencia de anticuerpos anti β2glicoproteína 1 dependientes de anticardiolipinas .
También se puede observar en pacientes con diagnóstico positivo trombocitopenia y anticuerpos anti β2-glicoproteína 1 (no dependiente de cardiolipinas) y anticuerpos anti fosfatidilserina.
Criterios
El diagnóstico de SAFL requiere tanto de evidencia clínica (eventos clínicos documentados tales como trombosis vascular o problemas obstétricos), como de la presencia confirmada de anticuerpos antifosfolípidos en ensayos repetidos. El criterio de clasificación de Sapporo para el SAFL (1998, publicado en 1999), fue reemplazado luego por el criterio de Sydney en 2006.
De acuerdo al criterio de clasificación más reciente, un diagnóstico de SAFL requiere una manifestación clínica y una prueba de laboratorio.
- Clínica:
- Un episodio documentado de trombosis arterial, venosa o de pequeños vasos que no sea trombosis venosa superficial en ningún tejido u órgano, y validada por un criterio objetivo sin evidencia significativa de inflamación en el vaso sanguíneo y/o:
- 1 o más muertes fetales inexplicables de un feto de al menos 10 semanas de gestación morfológicamente normal (documentado por medio de ultrasonografía o examen directo), y/o 3 o más abortos espontáneos consecutivos antes de las 10 semanas de gestación, habiendo descartado anormalidades anatómicas u hormonales de la madre y anormalidades cromosomales tanto maternas como paternas. O al menos 1 nacimiento prematuro de un neonato morfológicamente normal antes de las 34 semanas de gestación debido a eclampsia o preeclampsia severa de acuerdo a sus definiciones estándar, o evidencias reconocibles de insuficiencia placentaria mas
- Laboratorio:
- Anticardiolipinas IgG y/o IgM medida por un ensayo ELISA estandarizado y no dependiente de cofactores en 2 o más ocasiones, con no menos de 12 semanas de separación entre ambas, a títulos medios o elevados (p.ej. >40 GPL o MPL, o > percentilo 99) y/o
- Anti-β2 glicoproteína I IgG y/o IgM medida por un ensayo ELISA estandarizado en 2 o más ocasiones, con no menos de 12 semanas de separación; a títulos medios o elevados(> al percentilo) y/o
- Anticoagulante lúpico detectado en 2 ocasiones con no menos de 12 semanas de separación de acuerdo a las guías de la International Society of Thrombosis and Hemostasis.
Existen 3 entidades SAFL distintivas: el SAFL primario (en ausencia de cualquier comorbilidad), el SAFL secundario (cuando hay alguna condición autoinmune preexistentes, más frecuentemente el lupus eritematoso sistémico), y el SAFL catastrófico (cuando aparece falla multiorgánica simultánea con oclusión de pequeños vasos).
De acuerdo a las declaraciones del consenso de 2006, es conveniente, para propósitos de investigación, clasificar al SAFL en una de las siguientes categorías:
- I: más de un criterio de laboratorio presentes en cualquier combinación;
- IIa: presencia sólo de anticoagulante lúpico
- IIb: presencia sólo de anticardiolipinas IgG y/o IgM a títulos medios o altos
- IIc: presencia sólo de anti-β2 glicoproteína I IgG y/o IgM en títulos mayores al percentilo 99.
La Declaración de Consenso Internacional se utiliza comúnmente para el diagnóstico de SAFL catastrófico. Basado en estas declaraciones, el diagnóstico definitivo de SAFLC requiere:
- a) Trombosis vascular en tres o más órganos o tejidos y
- b) Desarrollo de manifestaciones secundarias simultáneamente o antes de la semana 'y
- c) Evidencia de trombosis de pequeños vasos en al menos un órgano o tejido y
- d) Confirmación de laboratorio para la presencia de anticuerpos antifosfolípidos.
Algunos ensayos serológicos para sífilis pueden dar resultados positivos en algunos pacientes con anticuerpos antifosfolípidos positivos (los aPL se unen inespecíficamete a los lípidos presentes en el ensayo y entregan un falso positivo) aunque los test más específicos para sífilis que utilizan antígenos recombinantes darán un resultado negativo.
El desarrollo en ei diagnóstico del síndrome de anticuerpos antifosfolipdos (SAFL) en los últimos años mostró que un perfil de anticuerpos puede ser útil para la diferrenciación clínica entre pacientes con SAFL y pacientes con trombosis venosa o arterial. Desde 2014 con un ensayo Immuno DOT para antifosfolipidos, que contiene diez diferentes fosfolipidos o proteínas de unión a fosfolipidos, hay una nueva posibilidad de medir el factor de riesgo de patientos con SAFL. A través de este ensayo es posible medir simultáneamente, gracias a una distribución optimizada de epitopos sobre una membrana hidrofóbica, anticardiolipinas, fosfatidilnositol, fosfatidilserina, fosfatidicolina, fosfatidiletanolamina, fosfatidilglicerol, ácido fosfatidico, anexina V y protrombina con una mayor sensibilidad que los 2 recomendados ensayos ELISA ("antibody profiling in APS" publicado en Lupus 2014 23 página 1262 en línea http://lup.sagepub.com/content/23/12/1262). Estoy resultados fueron confirmados para un estudio multicéntrico ("antiphospoholipid antibodies detected by line immunoassay differentiate among patients with phospholipid symdrome, with infections and asmptomatic carries" publicado en Arthritis Research & therapy (2016) 18:111) mostrando una notable asociación entre anticuerpos antifosfolipidos y la manifestación de SAFL general o asociado con complicationes espedeficas (aborto habitual o ataque cerebrovascular).
Tratamiento
Muy frecuentemente, esta enfermedad se trata prescribiendo aspirina para inhibir la activación plaquetaria o warfarina como anticoagulante. El objetivo del tratamiento profiláctico es mantener el INR del paciente entre 2,0 y 3,0. Sin embargo, no es frecuente hacerlo con pacientes que no presentan ninguna clase de síntomas trombóticos. Durante el embarazo se utilizan heparinas de bajo peso molecular y aspirina en bajas dosis en lugar de warfarina, debido a la teratogenicidad de ésta. A las mujeres que han sufrido abortos espontáneos recurrentes se les aconseja que comiencen a tomar aspirina e inicien el tratamiento con heparina de bajo peso molecular apenas reconozcan que desaparece su ciclo menstrual. En los casos refractarios se puede utilizar la plasmaféresis.[cita requerida]
Pronóstico
El pronóstico del SAFL a largo plazo se determina principalmente por la recurrencia de las trombosis, las cuales pueden ocurrir en más del 39% de los pacientes, algunas veces incluso bajo terapia antitrombótica.[cita requerida]
Historia
El síndrome antifosfolípido fue descrito completamente en los años 1980 tras varios informes previos acerca de anticuerpos específicos en personas con lupus eritematoso sistémico y trombosis. El síndrome es a veces denominado por su epónimo como "Síndrome Hughes", en honor al reumatólogo Dr. Graham R.V. Hughes (St. Thomas' Hospital, Londres, UK), quien trabajó en la Unidad de Lupus Louise Coote del Hospital St Thomas y que desempeñó un papel fundamental en la descripción del cuadro.
Véase también
- Anticuerpos antifosfolípidos
- Anticoagulante lúpico
- Anticuerpos anticardiolipinas
- Autoanticuerpos
- Síndrome de Susac
Bibliografía
- Triona Holden (2003). Positive Options for Antiphospholipid Syndrome (APS): Self-Help and Treatment. Hunter House (CA). ISBN 0-89793-409-1.
- Kay Thackray (2003). Sticky Blood Explained. Braiswick. ISBN 1-898030-77-4. Una apreciación personal sobre cómo lidiar con la condición.
- Graham R V Hughes (2009). Understanding Hughes Syndrome: Case Studies for Patients. Springer. ISBN 1-84800-375-7. 50 casos de estudio.
Enlaces externos
- APS Foundation of America, Inc.
- Hughes Syndrome Foundation (fundación del Síndrome de Huges)
- Entrevista con Hughes The Daily Telegraph 2 de febrero de 2009. Consultada 3 de febrero de 2009.
- Asociación española Síndrome Antifosfolípido