
| Osteogénesis imperfecta | ||
|---|---|---|
 Se trasmite de una forma autosómica dominante
| ||
| Especialidad | Pediatría, genética médica, ortopedia | |
| Síntomas | Huesos que se rompen con facilidad, tinte verde en la esclerótica (parte blanca del ojo), estatura baja, hipermovilidad articular , pérdida auditiva | |
| eMedicine | ped/1674 | |
| Sinónimos | ||
| enfermedad de los huesos frágiles; Fragilitas osio; enfermedad de Lobstein; IO; osteopsiatirosis; enfermedad de Porak y Durante; enfermedad de Vrolik | ||
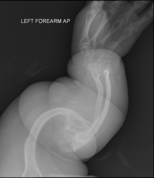


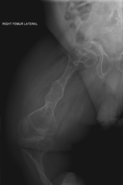
La osteogénesis imperfecta u osteogenia imperfecta (también llamada enfermedad de los huesos de cristal) es un trastorno congénito, es decir, presente al nacer, que se caracteriza por una fragilidad de hueso excesiva, como consecuencia de una deficiencia congénita en la elaboración de una proteína, el colágeno tipo I. Quienes portan el defecto tienen menos colágeno de lo normal o es de una menor calidad y como es una proteína importante en la estructura de los huesos, causa una fragilidad y debilidad poco usual de los huesos. El diagnóstico es radiológico, incluso antes del parto.
Causas
En la mayoría de casos la osteogénesis imperfecta es una enfermedad autosómica dominante debida a errores en el gen COL1A1 o COL1A2, lo que quiere decir que la persona la padecerá si tiene una copia del gen mutada. Una persona con osteogénesis imperfecta tiene un 50 % de posibilidades de transmitirle el gen y la enfermedad a sus hijos en este caso. Sin embargo, también puede deberse a errores en otros genes, como el CRTAP o el LEPRE1, los cuales siguen una herencia autosómica recesiva, es decir que sólo se manifiesta la enfermedad si el individuo lleva las dos copias del gen alteradas, por lo que sólo se transmite la enfermedad si ambos padres pasan una copia mutada del gen, cosa que puede suceder aunque ellos no padezcan la enfermedad. La mayoría de los casos de OI se heredan de los padres, aunque algunos casos son el resultado de nuevas mutaciones genéticas. El trastorno puede aparecer de nuevo por mutaciones esporádicas o por sus antepasados.
La penetrancia en los individuos heterocigotos para una mutación COL1A1 o COL1A2 es del 100%, aunque la expresión puede variar considerablemente, incluso en la misma familia.
Por lo general, se debe a la expresión defectuosa de las cadenas de procolágeno del tipo I. Existen muchos defectos diferentes que pueden afectar este gen y la gravedad de esta enfermedad depende del defecto específico de dicho gen. Por ejemplo, puede ocurrir debido a una mutación puntual de transversión (timina por guanina) en el procolágeno que impide la remoción de los péptidos terminales de la enzima procolágeno peptidasa.
En esta anormalidad ocurre el cambio de una Gly por una Cys en la posición 988* de la cadena Alfa I de la triple hélice, por esta razón se abre el extremo Ct a una excesiva hidroxilación y glicosilación que impide su corte enzimático y posterior ensamble durante la maduración del tropocolágeno, lo que se traduce en la aparición de fracturas a nivel del hueso.
Esta patología también se conoce como niños de cristal, en donde el colágeno I es almacenado, pero en el hígado.
Clasificación de Sillence
Actualmente, hay 17 tipos de OI diferentes clasificados. Aunque para la clasificación clínica, normalmente se utilizan solamente los cuatro primeros tipos. Aquí hay una tabla con los 17 tipos de OI registrados.
| Tipos | Características |
| I | Enfermedad de Ekman Lobstein, autosómica dominante. Se caracteriza por osteoporosis generalizada con fragilidad ósea anormal, escleróticas azules toda la vida, hay una reducción del espesor escleral y la uvea pigmentada subyacente se hace visible a través de la esclerótica adelgazada, pérdida de la audición de tipo conductivo presenil, la que puede ser de conducción por otoesclerosis o tipo nervioso por la compresión del nervio VIII al salir del cráneo y la otoesclerosis es resultado de la proliferación anormal del cartílago, el cual al calcificarse produce esclerosis de la porción petrosa del temporal. No todos los pacientes presentan dentinogénesis imperfecta. |
| II | Enfermedad de Vrolik, autosómica recesiva. Se caracteriza por fragilidad ósea extraordinaria, culmina en la muerte en el periodo perinatal o comienzos de la lactancia, huesos largos muy fragmentados (fémur en acordeón), prominencia de hueso parietal y temporal con occipucio colgante y osificación de cráneo retrasada extraordinariamente. |
| III | Autosómica dominante. Caracterizada por fragilidad ósea intensa ocasionando múltiples fracturas, deformidad progresiva de huesos largos, retardo grave del crecimiento con la talla más pequeña de todas las OI, escleróticas azules en el neonato, pero con la edad se vuelven menos azules, Dentinogénesis imperfecta, deformidad de columna por combinación de osteoporosis intensa, fracturas por compresión de vértebras e hiperlaxitud ligamentosa, cifoescoliosis, siendo la más común la escoliosis dorsal, cara aspecto triangular de “duende”, frente amplia, prominencia de huesos parietal, temporal y con occipucio colgante. |
| IV | Autosómica dominante. En el neonato las escleras tienen color normal, pueden llegar a estar azulados, sin embargo, se vuelven cada vez menos azules, osteoporosis, fragilidad ósea y deformidad de huesos largos, si presenta dentinogénesis imperfecta se clasifica en la subdivisión 4 A, si no presenta dentinogénesis imperfecta se clasifica 4 B. En la dentinogénesis imperfecta hay alteraciones por deficiencia de dentina, manchas pardo amarillentas o azuladas grisáceas translúcidas y los incisivos inferiores son los más afectados. |
| V | Autosómica dominante. Es similar al tipo 4. Es más común que se presente en niños que muestran escleras blancas, formación de callo hiperplásico, más común en fémur, tibia y húmero. En radiografías se advierte la formación masiva de callos en forma de mariposa, calcificación de la membrana interósea en el antebrazo y en consecuencia problemas en la pronosupinación de miembros superiores, dislocación de la cabeza radial anterior. |
| VI | Autosómica recesiva. Fenotipo de moderado a severo, se presentan fracturas en los dos primeros años de vida, escleróticas normales o azul claro, fracturas de vértebras, en este tipo no hay dentinogénesis imperfecta. |
| VII | Se parece a los tipos V i II. Tala baja y acortamiento de extremidades. Es un tipo de OI recesivo que se debe a una mutación en el gen CRTAP. |
| VIII | Se parece a los tipos II y III. Grave deficiencia de crecimiento y extrema desmineralización del esqueleto. OI recesiva debida a una mutación en el gen LEPRE1. |
| IX | Manifestación clínica muy variable (de moderada a muy grave). Se debe a una mutación en el gen PPIB. Es una OI de herencia recesiva. |
| X | Manifestación clínica de grave a letal. Se debe a una mutación en el gen SERPINH1. Es una OI de herencia recesiva. |
| XI | Es un tipo de OI progresivo que puede cursar con deformaciones de la columna vertebral. También se llama síndrome de Bruck I. Su herencia es recesiva. Se debe a una mutación en el gen FKBP10. |
| XII | Es una OI con una manifestación clínica de moderada a grave, cuyas características incluyen fracturas frecuentes, ligeras deformidades óseas y erupción dental retrasada. No está asociada a ningún problema auditivo y la esclerótica de los afectados es de color normal. Se debe a mutaciones en el gen SP7/OSX. |
| XIII | Se trata de una OI causada por mutación recesiva en la proteína morfogenética ósea 1(BMP1). El fenotipo es grave y puede presentar deformaciones óseas generalizadas, múltiples fracturas, falta de remodelación ósea y reducción generalizada de la densidad ósea. |
| XIV | El tipo XIV es una OI entre moderada y grave debida a mutaciones recesivas en el gen TMEM38B. Los afectados tienen osteopenia y fracturas, pero no presentan escleróticas azuladas ni dentinogénesis imperfecta. Tampoco tienen pérdida de audición ni ninguna otra afectación orgánica. |
| XV | La OI tipo XV se debe a mutaciones recesivas en el gen WNT1. Su manifestación clínica es variable, de moderada a progresivamente deformante. Los afectados pueden tener la esclerótica azulada, pero tanto los dientes como la audición son normales. Este tipo de OI puede estar asociado a anomalías cerebrales. Las características clínicas incluyen las fracturas frecuentes, deformidades óseas, compresiones vertebrales graves, importante reducción de la densidad ósea y baja estatura. |
| XVI | Se debe a una alteración genética del transductor OASIS (codificado en el gen CREB3L1). Los afectados tienen una OI grave. Los portadores heterocigóticos de la mutación (progenitores no afectados) poseen ciertos rasgos de la OI tipo I, pero sin fracturas. |
| XVII | Se debe a una mutación en el gen SPARC. |
Tratamiento
La osteogénesis imperfecta no tiene cura y para mantener un estilo de vida saludable, hay que realizar ejercicio para ayudar a prevenir fracturas. El tratamiento puede incluir analgésicos, fisioterapia, aparatos ortopédicos o sillas de ruedas y cirugía (colocando varillas de metal a través de huesos largos para fortalecerlos).
Bifosfonatos
En la OI grave, el pamidronato reduce el dolor óseo, evita nuevas fracturas vertebrales, reformula cuerpos vertebrales previamente fracturados y reduce el número de fracturas de huesos largos. Aunque los bifosfonatos orales son más convenientes y más baratos, no se absorben tan bien, y los bifosfonatos intravenosos son generalmente más efectivos, aunque esto está en estudio. Algunos estudios han encontrado bisfosfonatos orales e intravenosos, como alendronato oral y pamidronato intravenoso, equivalentes. En un ensayo con niños con OI leve, el risedronato oral aumentó la densidad mineral ósea y redujo las fracturas no vertebrales.
Cirugía
Las varillas de metal se pueden insertar quirúrgicamente en los huesos largos para mejorar la fuerza. La colocación de varillas de acero inoxidable en los canales intramedulares de los huesos largos los estabiliza y fortalece. Su tratamiento es útil con la rehabilitación, ya que previene fracturas y forma parte de la base para el tratamiento ortopédico. La fusión espinal se puede realizar para corregir la escoliosis, aunque la fragilidad ósea inherente hace que esta operación sea más compleja en los pacientes. La cirugía para impresiones basilares puede llevar a cabo un cabo si la presión ejercida sobre la médula espinal y el tronco encefálico está causando problemas neurológicos.
Fisioterapia
La fisioterapia se usa para fortalecer los músculos y mejorar la motilidad de una manera suave, mientras se minimiza el riesgo de fractura. Esto a menudo implica hidroterapia, ejercicios de resistencia ligera y el uso de cojines de apoyo para mejorar la postura. Se alienta a las personas a cambiar de posición regularmente durante el día para equilibrar los músculos que se usan y los huesos bajo presión.
Enlaces externos
-
 Wikimedia Commons alberga una categoría multimedia sobre Osteogénesis imperfecta.
Wikimedia Commons alberga una categoría multimedia sobre Osteogénesis imperfecta. - Asociación de Huesos de Cristal de España (AHUCE). Osteogénesis Imperfecta(OI)
- Fundación AHUCE
- Longo DL, Kasper DL, Jameson JL, Fauci AS, et al. Harrison Principios de Medicina Interna. 18a ed. McGraw Hill; 2012.ISBN 978-0071748896
- Gutierrez-Diez MP, Molina Gutiérrez MA, Prieto L, Parra JI, et al. Osteogénesis Imperfecta: Nuevas Perspectivas. Revista Española Endocrinología Pediátrica 2013; 4 (Suppl) 10.3266.
| Control de autoridades |
|
|---|
-
 Datos: Q749409
Datos: Q749409
-
Multimedia: Osteogenesis imperfecta / Q749409