

El NF-kB (factor nuclear potenciador de las cadenas ligeras kappa de las células B activadas) es un complejo proteico que controla la transcripción del ADN. NF-kB se encuentra en la mayoría de tipos de células animales y está implicado en la respuesta celular frente a estímulos como el estrés, las citoquinas, radiación ultravioleta, LDL oxidadas y antígenos bacterianos o virales. El NF-κB juega un papel clave en la regulación de la respuesta inmune debida a la infección (las cadenas ligeras kappa son componentes cruciales de las inmunoglobulinas). La regulación defectuosa del NF-kB está relacionada con el cáncer, enfermedades inflamatorias y autoinmunes, shock séptico, infecciones virales o un desarrollo inmune inadecuado. También está implicado en procesos de plasticidad sináptica y de memoria.
Descubrimiento
El NF-κB fue descubierto en el laboratorio por el galardonado con el Premio Nobel David Baltimore a partir de su interacción con una secuencia de 11 pares de bases en la región potenciadora (enhancer) de la cadena ligera kappa de la inmunoglobulina en células B.
Estructura
Todas las proteínas de la familia de los NF-κB comparten un dominio de homología Rel en su extremo N-terminal. Una subfamilia de proteínas NF-κB, incluidas RelA, RelB y c-Rel, tienen un dominio de transactivación en su extremo C-terminal. En contraste, las proteínas NF-κB1 y NF-κB2 son sintetizadas como precursores, p105 y p100, que tras madurar dan lugar a las subunidades de NF-κB, p50 y p52 respectivamente. La maduración de p105 y p100 está mediada por la vía ubiquitina/proteosoma e implica la degradación selectiva del C-terminal de la región que contiene repeticiones de anquirina. Mientras que la formación de p52 a partir de p100 es un proceso altamente regulado, la p50 se forma por un proceso constitutivo de la p105.
Miembros
Los miembros de la familia del NF-κB comparten estructura homóloga con la oncoproteína retroviral v-Rel, lo que da lugar a su clasificación como proteínas NF-κB/Rel.
En mamíferos existen 5 miembros de la familia NF-κB:
| Clase | Proteína | Alias | Gen |
|---|---|---|---|
| I | NF-κB1 | p105 → p50 | NFKB1 |
| NF-κB2 | p100 → p52 | NFKB2 | |
| II | RelA | p65 | RELA |
| RelB | RELB | ||
| c-Rel | REL |
Distribución de especies y evolución
Además de en mamíferos, el NF-κB se encuentra también en animales más sencillos. Entre ellos los cnidarios (como las anémonas de mar, el coral o la hidra), poríferos (esponjas), el parásito unicelular eucariota Capsaspora owczarzaki e insectos (como polillas, mosquitos y moscas de la fruta). La sucesión de los genomas de los mosquitos A. aegypti y A. gambiae, y la mosca de la fruta D. melanogaster ha permitido realizar estudios comparativos genéticos y evolutivos de NF-κB. En estas especies de insectos, la activación de NF-κB está provocada por la vía Toll (que evolucionó de forma independiente en insectos y mamíferos) y por la vía IMD (inmunodeficiencia).
Señalización
Activación

El NF-κB es relevante en la regulación de la respuesta celular ya que pertenece a la categoría de los factores de transcripción primarios de “acción rápida”, como son los factores de transcripción que están presentes en las células en un estado de inactivación y que no requieren una nueva síntesis de proteínas para ser activados (otros miembros de esta familia incluyen factores de transcripción tales como c-Jun, STATs, y receptores nucleares hormonales. Esto permite al NF-κB ser la primera respuesta a estímulos celulares nocivos. Los inductores de la actividad del NF-κB son altamente variables, y pueden ser desde especies reactivas de oxígeno (ROS), factores de necrosis tumoral alfa (TNF α), interleucina 1-beta (IL-1β), lipopolisacáridos bacteriales (LPS), isoproterenol, cocaína e incluso radiaciones iónicas.
El receptor activador del factor nuclear kappa B (RANK), que es un tipo de TNFR, es un activador central del NF-κB. La osteoprotegerina (OPG), que es un receptor señuelo homólogo al ligando de RANK, inhibe RANK uniéndose a RANKL, y así, la osteoprotegerina está estrechamente relacionada con la regulación de la activación de NF-κB.
Muchos productos bacteriales así como la estimulación de una gran variedad de receptores de la superficie celular inducen la activación de NF-κB así como también cambios rápidos en la expresión génica. La identificación de receptores de tipo Toll (TLRs) como patrones específicos de reconocimiento molecular, y el descubrimiento que la estimulación de TLRs conduce a la activación de NF-κB, mejoró la comprensión de como los diferentes patógenos pueden llegar a activar NF-κB. Por ejemplo, diversos estudios han identificado TLR4 como un receptor para el componente LPS de las bacterias Gram-Negativas. Los TLRs son reguladores esenciales tanto de las respuestas inmunes innatas como de las adaptativas.
A diferencia de RelA, RelB, y c-Rel, las subunidades p50 y p52 de NF-κB no contienen dominios de transactivación en sus mitades C-terminales. No obstante, las p50 y p52 de NF-κB juegan un papel importante modulando la especificidad de la función de NF-κ . Aunque los homodímeros de p50 y p52 son, en general, represores del lugar de transcripción de κB, ambos (p50 y p52) participan en la transactivación de genes diana formando heterodímeros con RelA, RelB, o c-Rel. Además, los homodímeros p50 y p52 también se unen a la proteína nuclear Bcl-3, pudiendo dichos complejos funcionar como activadores transcripcionales.
Inhibición
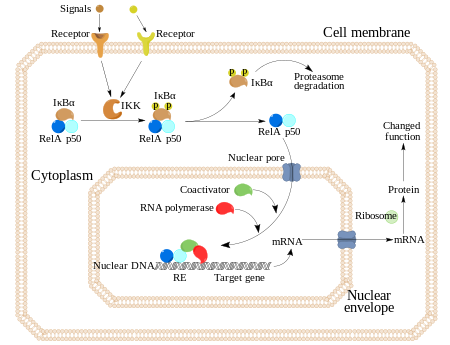
En células no estimuladas, los dímeros de NF-κB son secuestrados en el citoplasma por una familia de inhibidores, llamados IκBs (Inhibidores de κB), los cuales son proteínas que contienen múltiples copias de una secuencia llamada “repeticiones de anquirina”. En virtud de estos dominios, las proteínas IκB enmascaran la secuencia de localización nuclear (NLS) de las proteínas NF-κB y las mantienen secuestradas en un estado de inactivación en el citoplasma.
Las IκBs forman una familia de proteínas relacionadas, que tienen un dominio N-terminal regulador, seguido por seis o más repeticiones de anquirina y un dominio PEST cerca de su C-terminal. Aunque la familia de las IκB consiste en IκBα, IκBβ, IκBε, y Bcl-3, la principal y más estudiada IκB es la IκBα. Debido a la presencia de repeticiones de anquirina en las mitades C-terminales, p105 y p100 también funcionan como proteínas IκB. La mitad C-terminal de p100, que a menudo es conocida como IκBδ, también funciona como inhibidor. La degradación de IκBδ en respuesta a estímulos de desarrollo, tales como aquellos transducidos a través de LTβR, potencia la activación del dímero NF-κB a través de una vía NIK dependiente no canónica.
La activación de NF-κB se inicia a través de la degradación inducida por señal de proteínas IκB. Esto ocurre principalmente mediante la activación de una quinasa llamada IκB quinasa (IKK). La IKK está formada por un heterodímero de las subunidades catalíticas de IKK alfa e IKK beta y por una proteína reguladora “maestra” llamada NEMO (modulador esencial NF-κB) o IKK gamma. Cuando es activada por señales, normalmente procedentes de fuera de la célula, la IκB quinasa fosforila dos residuos de serina localizados en un dominio IκB regulador. Una vez fosforiladas estas serinas (por ejemplo, las serinas 32 y 36 en la IκBα humana), las moléculas del inhibidor IκB son modificadas por un proceso llamado ubiquitinación, que después las lleva a ser degradadas por una estructura celular llamada proteosoma.
Con la degradación de IκB, el complejo NF-κB es libre para entrar al núcleo dónde puede activar la expresión de los genes específicos que tienen cerca sitios de unión de ADN para NFκB. La activación de estos genes por NF-κB entonces induce una respuesta fisiológica, como por ejemplo, una respuesta inflamatoria o inmune, una respuesta de supervivencia celular, o una proliferación celular. NFκB activa la expresión de su propio represor, IκBα. El nuevamente sintetizado IκBα re-inhibe NFκB y, por tanto, forma un bucle de auto feedback, que provoca niveles oscilatorios de la actividad de NFκB. Además, varios virus, incluyendo el virus VIH del SIDA, poseen sitios de unión para NFκB que controlan la expresión de genes virales, que a su vez contribuyen a la replicación viral o a la patogenicidad viral. En el caso de VIH-1, la activación de NFκB puede verse involucrada, al menos en parte, en la activación del virus desde un estado latente e inactivo. YopP es un factor secretado por la Yersinia pestis, el agente causante de la peste, que previene la ubiquitinación de IκB. Esto provoca que este agente patógeno inhiba eficazmente la vía NF- κB y así bloquea la respuesta inmune de una persona infectada con Yersinia.
Inhibidores de la actividad NF-κB
En cuanto a las proteínas inhibidoras de la actividad NF-κB, una de ellas, IFRD1, reprime la actividad de la p65 de NF-κB mediante la mejora de la desacetilación mediada por HDAC de la subunidad p65 a lisina 310, favoreciendo el secuestro de la HDAC3 a p65. De hecho, IFRD1 forma complejos trimoleculares con p65 y HDAC3.
No canónico
Un selecto grupo de estímulos de diferenciación celular o desarrollo celular, tales como linfotoxina-α, BAFF o RANKL, activan la vía no canónica de NF-κB para inducir el dímero NF-κB/RelB:p52 en el núcleo. En esta vía, la activación de la quinasa inductora de NF-κB (NIK) sobre el ligando del receptor induce la fosforilación y el posterior procesamiento proteosomal de la proteína precursora de NF-κB2 de p100 a subunidad p52 madura a través de una vía IKK1/IKKa dependiente. Entonces la p52 dimeriza con RelB para aparecer como RelB:p52 nuclear con actividad de unión al ADN para regular una clase distinta de genes. En contraste con la señalización canónica, que responde a la degradación mediada por NEMO-IKK2 de IκBα, -β, -ε, la señalización no canónica difícilmente depende del proceso de p100 a p52 mediado por NIK. Teniendo en cuenta sus diferentes regulaciones, se pensaba que estas dos vías eran independientes una de otra. No obstante, recientes análisis han revelado que la síntesis de los constituyentes de la vía no canónica, es decir RelB y p52, es controlada por la señalización canónica de IKK2-IκB-RelA:p50. Por otra parte, la generación de los dímeros tanto canónicos como no canónicos, es decir RelA:p50 y RelB:p52, en el medio celular, también están relacionadas entre sí de manera mecánica. Estos análisis sugieren que un sistema NF-κB integrado conduce a la activación de los dímeros RelA y RelB y que un mal funcionamiento de la vía canónica puede dar lugar a una respuesta celular aberrante también a través de la vía no canónica.
Inmunidad
El NF-κB2 es un factor de transcripción principal que regula genes responsables tanto de la respuesta innata como de la respuesta adaptativa. Tras la activación de los receptores de los linfocitos T o B, el factor NF-κB se activa a través de distintos componentes de señalización. El receptor de la célula T, la proteína quinasa Lck, es reclutada y fosforilada por los motivos ITAM que se encuentran en el citoplasma de CD3. A continuación la molécula ZAP es reclutada y fosforilada por ITAM que ayudan a recluir LAT y PLC gamma, las cuales causan la activación de PKC: a través de una cascada de eventos de fosforilación, el complejo quinasa se activa y NF-κB es capaz de entrar en el núcleo para regular al azar los genes implicados en el desarrollo, maduración y proliferación de las células T.
En neuronas
Además de las funciones en la supervivencia celular, el NF-κB tiene diversas funciones en el sistema nervioso, incluyendo la plasticidad, el aprendizaje y la memoria neuronal. Además de los estímulos que activan el NF-κB en otros tejidos, en el sistema nervioso puede ser activado por factores de Crecimiento (BDNF, NGF) utilizando el glutamato como neurotransmisor. Estos activadores de NF-κB en el sistema nervioso convergen en el complejo de IKK y la vía canónica.
Recientemente se ha mostrado un gran interés por el papel de NF-κB en el sistema nervioso. Los estudios actuales sugieren que el NF-κB es importante para el aprendizaje y la memoria en múltiples organismos como cangrejos, moscas de la fruta, y ratones. El factor NF-κB puede regular el aprendizaje y la memoria, en parte, por la modulación de la plasticidad, función sináptica, así como el crecimiento de las dendritas y de las espinas dendríticas.
Se ha demostrado que los genes que disponen de sitios de unión para el NF-κB, tienen un mayor aprendizaje en la siguiente transcripción genética,[8] lo que sugiere que este factor en el sistema nervioso es importante para la plasticidad y el aprendizaje. Muchos de estos genes diana de NF-κB son los receptores de glutamato (AMPA y NMDA-R-R), los factores de crecimiento (BDNF, NGF), citoquinas (TNF-alfa, TNFR), quinasas (PKAc), y las proteínas de unión sináptica (PSD-95).
Importancia clínica
Descripción general de las vías de transducción de señales implicadas en la apoptosis. El NF-κB es ampliamente utilizado por las células eucariotas como regulador de los genes que controlan la proliferación celular y la supervivencia celular. Debido a ello, muchos tipos diferentes de tumores humanos tienen mal regulados el NF-κB (es decir, activado). Cuando NF-κB se encuentra activado, induce la expresión de los genes que protegen e inducen la proliferación de células que en otras condiciones, deberían morir por apoptosis, para evitar que el daño se extienda.
Los defectos en NF-κB producen una mayor susceptibilidad a la apoptosis que conduce que haya un aumento en la muerte celular . Esto se debe a que NF-κB regula genes anti-apoptóticos (sobre todo la TRAF1 TRAF2) y por tanto, controla la actividad enzimática de las caspasas, que son fundamentales para la mayoría de los procesos apoptóticos.
En las células tumorales, NF-κB se activa, ya sea debido a mutaciones en genes que codifican los factores de transcripción de NF-κB o en los genes que controlan la actividad de NF-κB (por ejemplo, genes IkB) y, además, algunas células tumorales secretan factores que son activadores de NF -kB. El bloqueo de NF-κB puede causar que las células tumorales dejen de proliferar, morir, o que sean más sensibles a la acción de los agentes anti-tumorales. Por lo tanto, NF-κB es el objeto de muchas empresas farmacéuticas para la terapia contra el cáncer.
Debido a que NF-κB controla varios genes involucrados en la inflamación, no es de extrañar que NF-κB se encuentre activado crónicamente en enfermedades inflamatorias, tales como la enfermedad inflamatoria intestinal, la artritis, sepsis, gastritis, asma y arterosclerosis entre otros. Es importante señalar que los reguladores clave de la NF-κB se asocian con una elevada mortalidad, especialmente en enfermedades cardiovasculares y esquizofrénicas.
Muchos productos naturales , incluidos los anti-oxidantes que tienen actividad anticancerígena y antiinflamatoria , se ha demostrado que también son capaces de inhibir NF-κB. Hay una polémica patente en los EE.UU que aplica el descubrimiento y el uso de agentes que pueden bloquear la NF-κB con fines terapéuticos. Esta patente está involucrada en varios juicios, incluyendo Ariad v Lilly. Un trabajo reciente de Karin, Ben-Neriah y otros han remarcado la importancia de la conexión entre el NF-κB, la inflamación y el cáncer, dando una mayor importancia a las terapias inhibitorias de NF-κB.
Extractos procedentes de un gran número de plantas también son inhibidores de la activación de NF-κB in vitro.
Área terapéutica
Se ha observado que en muchos cánceres hay una sobre activación del factor NF-κB. Por otra parte, la supresión de NF-κB limita la proliferación de células cancerosas. Además, NF-κB juega un papel clave en la respuesta inflamatoria. Por lo tanto, los métodos de inhibición de señalización de NF-κB tienen gran interés terapéutico en cánceres y enfermedades inflamatorias.
La activación de la translocación nuclear de NF-κB es independiente al aumento de estrés oxidativo, por lo que, gracias a ello, se puede llevar a cabo un mejor estudio de posibles estrategias mediante inhibición de NF-κB.
Un nuevo medicamento llamado denosumab actúa aumentando la densidad mineral ósea y, por lo tanto, reduciendo las fracturas óseas en muchos pacientes. Inhibe la RANKL, que actúa a través de su receptor RANK, que a su vez promueve la NF-κB, RANKL normalmente permite la diferenciación de los osteoclastos a partir de monocitos.
Otros medicamentos son el disulfiram, olmesartán y ditiocarbamatos, que inhiben la cascada de señalización del factor nuclear κB (NF-κB).
Carcinoma Nasofaríngeo, EBV y NFKB
El Carcinoma Nasofaríngeo (NPC) se caracteriza por células epiteliales tumorales poco diferenciadas que residen en la parte posterior a la faringe. En este cuadro clínico se produce un microambiente complejo con gran infiltración de linfocitos, dando lugar a la aparición de un linfoepitelioma con un aparente fenotipo inflamatorio. Además en un 90% de los casos las células malignas son uniformemente positivas para el virus de Epstein-Barr (VEB), un herpes virus muy extendido en la población humana.
Estudios funcionales y genómicos recientes han implicado la activación de la vía NF-κB y la evasión inmunitaria en este tipo de carcinoma nasofaringeo asociados a EBV. En estos se muestran una activación constitutiva de las vías inflamatorias de NF-κB en un 90% de los casos que padecen Carcinoma Nasofaríngeo (NPC) asociados al virus de Epstein-Barr, ya sea a través de alteraciones somáticas o a través de la expresión del oncogén LMP1 viral codificado por el virus. Esta señalización aberrante de NF-κB ayuda a la progresión de este tipo de cáncer asociado a EBV, decrito como una característica genómica. En cuanto a la evasión de sistema inmune, se ha decrito que el 91,4% de los casos de carcinoma Nasofaringeo positivo para EBV, padece de alteraciones somáticas o sobreexpresión de varios genes virales (LMP1, BNLF2a) dirigidos a la inmunidad innata y adaptativa. Es probable que esta característica surja para contrarrestar el entorno inflamatorio debido a la infección persistente por el virus.
Véase también
Enlaces externos
- MeSH: NF-kappa+B (en inglés)
- Sankar Ghosh (2006). Handbook of Transcription Factor NF-κB. Boca Raton: CRC. ISBN 0-8493-2794-6.
- Thomas D Gilmore. «The Rel/NF-κB Signal Transduction Pathway». Boston University. Consultado el 2 de diciembre de 2007.
- Traducción de en:NF-κB (versión: http://en.wikipedia.org/wiki/NF-%CE%BAB)
| Control de autoridades |
|
|---|
-
 Datos: Q411114
Datos: Q411114
-
 Multimedia: NF-kappa B / Q411114
Multimedia: NF-kappa B / Q411114