
| Leucemia mieloide crónica | ||
|---|---|---|
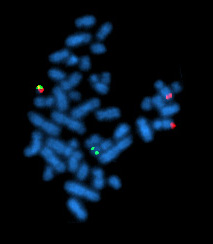 El cromosoma Filadelfia, tal como se ve durante la metafase mediante FISH.
| ||
| Especialidad | oncología | |
| Sinónimos | ||
|
leucemia mielógena crónica. Leucemia granulocítica crónica. Leucemia mielocítica crónica | ||
La leucemia mieloide crónica (LMC) catalogada como cáncer, es una neoplasia mieloproliferativa crónica que afecta a las células precursoras o hemocitoblastos de la sangre. Se caracteriza por una proliferación de los glóbulos blancos de la serie granulocítica, esplenomegalia, trombocitosis y anemia. Se origina por una mutación genética, la traslocación entre los cromosomas 9 y 22.
Epidemiología
- Incidencia: 1 a 2 casos nuevos por cada 100 000 habitantes al año.
- Variación entre géneros: razón varón/mujer de 2,0/1,2 (EE. UU., 1999).
- Variación con la edad: aumento progresivo hasta los 45 años, luego aumento mayor.
- Progresión temporal: disminución leve de la incidencia (1,9 nuevos casos / 100 000 habitantes al año; EE. UU., 1973).
Causas
La causa de la LMC es desconocida. No hay evidencia de que tenga relación con fármacos o infecciones; los estudios de los efectos de las bombas atómicas y de los supervivientes del accidente nuclear de Chernobil demuestran que solo grandes dosis de radiación pueden inducir la aparición de una LMC.
Lo que sí se conoce es que aparece una traslocación genética de tipo t(9;22) que produce un reordenamiento de los genes BCR/ABL, produciendo el denominado cromosoma Filadelfia descubierto en 1960 por Newell y Hungerford. La proteína que resulta es una tirosina quinasa cuya alteración transforma el ATP en ADP, fosforilando un sustrato que altera la médula ósea y su funcionamiento.
Patogenia
Se produce la fusión entre el gen Abelson de la leucemia murina (ABL1) del cromosoma 9 con el gen BCR del cromosoma 22, conocido como cromosoma Filadelfia. Esto resulta en la expresión de la oncoproteína BCR-ABL1 quien es una proteína quinasa, que interviene por medio de la fosforilación descontrolada de los intermediarios implicados en la división celular, conduciendo a una proliferación infinita de las células mieloide.
Anatomía patológica

La LMC puede afectar diferentes órganos:

Sangre: Hay un elevado recuento leucocitario y es característico que sobrepase los 20 000 blancos/mm3 pudiendo alcanzar cifras de 200 000 blancos/mm3. Células granulocíticas en vías de maduración: Las más numerosas son mielocitos y neutrófilos. Puede encontrarse un elevado número de eosinófilos y basófilos circulantes así como de monocitos. Al inicio puede haber moderada trombocitosis.
Bioquímicamente es típico que los valores de Fosfatasa alcalina leucocitaria estén reducidos y su aumento puede indicar una reacción leucemoide por infección o que la LMC pasó a Leucemia mieloide aguda (hecho que ocurre con frecuencia).
Médula ósea: Macroscópicamente: En cortes de huesos de pacientes con LMC (cresta ilíaca) la médula ósea es reemplazada por un tejido blando color rosa pálido o marrón verduzco que refleja focos de hemorragia reciente que se extienden hacia todo el hueso.
Microscópicamente: Puede observarse panhiperplasia que ocupa entre 80-95% de las celdillas, con proliferación predominante de la serie granulocítica. La relación mieloeritroide varía entre 10:1 y 50:1 (lo normal es de 2:1 a 4,5:1). Cuando pasa a la fase blástica se puede evidenciar: 60% de mielocitos, 20% de linfocitos y 20% de eritrocitos y megacariocitos. Además hay un elevado número de eosinófilos, basófilos y monocitos, así como megacariocitos numerosos con falta de aglomeración y atipias. Hay fragmentos de núcleos megacariocíticos en sangre. La proporción de linfocitos es reducida tanto en médula ósea como en sangre periférica. Existe fibrosis de médula ósea infrecuente y se observa en los estadios terminales.
Bazo:
Macroscópicamente: La superficie de corte del bazo muestra expansión homogénea de la pulpa roja, con un aspecto moteado y áreas de infarto, así como la desaparición de los cuerpos de Malpighi.
Microscópicamente: Se observa metaplasma mieloide de los sinusoides y en forma menos predominante de los cordones. Existe predominio en la proliferación de células granulocíticas inmaduras. También se ven islotes de normoblastos eritroides y megacariocitos en cantidad variable.
Hígado: Se observa infiltración difusa, en algunos casos masiva, por células de LMC dando lugar a hepatomegalia moderada o severa que puede o no acompañarse de metaplasia mieloide la cual puede disponerse periportal, venacentrolobulillar y algunas veces con patrón perisinusoidal.
Ganglios linfáticos: Se observan afectados en etapas tardías de la enfermedad en donde se puede observar un infiltrado de células leucémicas que distorsiona la arquitectura normal del ganglio, que lo permeabiliza en las áreas persisinusoidales no viéndose los centros germinales.
Sistema nervioso central: Pueden verse infartos hemorrágicos como resultado de la oclusión microvascular difusa por agregados de células leucémicas.
Riñón: Se observa infiltración de células leucémicas que comienzan como pequeños agregados perivasculares que se extienden de forma progresiva a la totalidad del estroma. Pueden observarse cambios grasos y alteraciones en la bioquímica del ácido úrico.
Cuadro clínico
Tiene tres fases:
Fase crónica o mielocitaria
- Dura unos 4 a 5 años, aunque puede precederse de una fase previa asintomática, caracterizada solo por la alteración genética.
- Puede ser asintomática y detectarse en pruebas analíticas rutinarias, o presentar los siguientes síntomas:
- - Síntomas de hipoxia tisular (astenia, decaimiento, palidez, pérdida de peso, ...) resultantes de la hiperviscosidad producida por el aumento de la masa celular total de la sangre.
- - Síntomas derivados de la esplenomegalia: pesadez postprandrial, la saciedad precoz o fenómenos compresivos abdominales (típicamente en el hipocondrio izquierdo). Está en relación con las cifras leucocitarias, pero suele detectarse de forma más precoz.
- - Síntomas de hipercatabolismo celular (generalmente solo en casos más avanzados): hiperuricemia, hiperkalemia, insuficiencia renal.
- - No suele haber adenomegalias (no existen granulocitos en los ganglios linfáticos).
- El 80 - 85% de los pacientes son diagnosticados en esta fase.
- En sangre periférica se ve leucocitosis, a base de granulocitos, con menos de un 2% de blastos y en médula ósea proliferación de granulocitos, con disminución del tamaño de los precursores eritroides y megacariocíticos.
Fase acelerada
- Dura unos 6 u 8 meses.
- No se conocen bien los factores que promueven la transición a las siguientes fases de la enfermedad, pero los estudios citogenéticos y moleculares muestran nuevas alteraciones: la aparición de un segundo cromosoma Filadelfia, de una trisomía del cromosoma 8 o de una deleción p17-.
- El enfermo presenta fiebre, aumento de la anemia y sus consecuencias, dolores óseos, etc.
- En las pruebas analíticas aparece aumento de los basófilos (por aumento de blastos), hipereosinofilia, anemia y trombocitopenia. Como consecuencia, aparecen infecciones, trombosis y/o hemorragias.
Fase de transformación a leucemia aguda (crisis blástica)
- Aparecen más de un >20% de blastos en médula ósea. Por alteración genética de la célula madre en estadios madurativos más precoces, la leucemia mieloide crónica da crisis clínicas similares a la leucemia aguda. El 80% de los casos evolucionan a leucemia mieloblástica aguda (LMA), y el 20% a leucemia linfoblástica aguda (LLA), con mejor pronóstico.
- La clínica es de curso tormentoso, con anemia severa, infecciones de repetición, hemorragias y trombos, alteraciones multiorgánicas por infiltración linfocítica, signos de leucostasia, etc.
- La clínica es indistinguible de la de la leucemia aguda, y hay que hacer el diagnóstico diferencial por técnicas de biología molecular. La proteína resultante del gen híbrido presenta diferentes tamaños según la patología (190 KDa en la LLA; 210 KDa en la crisis blástica de la LMC; y 230 KDa en un tipo de síndrome mieloproliferativo crónico más infrecuente, la leucemia granulocítica crónica).
Diagnóstico
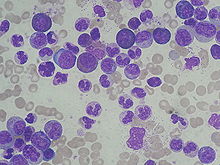

- Historia clínica: anamnesis y exploración. Datos de leucocitosis, anemia, trombocitopenia y esplenomegalia.
- Pruebas analíticas:
- Hemograma con leucocitosis (basofilia y eosinofilia), anemia y trombocitopenia variables según la gravedad, eritroblastos y algún blasto.
- Bioquímica con aumento de los niveles de LDH, hiperuricemia y disminución de los niveles de fosfatasa alcalina granulocítica (FAG).
- Biopsia de médula ósea: hipercelular.
- Citogenética: cromosoma Filadelfia; t(9;22) (en un 95% de los casos). A partir de la fase acelerada se pueden detectar, asimismo, trisomías 8 y 9.
- Técnicas de biología molecular que muestra el reordenamiento de los genes BCR/ABL.
Diagnóstico diferencial
Hay veces que para diagnosticar la LMC es suficiente cuando el paciente cursa con importante leucocitosis y significativa esplenomegalia. Pero otras veces cuando el paciente tiene una leucocitosis que es moderada es importante hacer el diagnóstico diferencial de LMC con otros síndromes mieloproliferativos, con leucocitos reactivos, infecciones, corticoterapia, con tumores metastásicos en médula ósea, estados de shock hemolisis o hemorragias agudas y reacción medular de agranulocitosis.
Los datos útiles para hacer el diagnóstico diferencial de LMC con otras patologías son: principalmente el análisis de biopsia ósea, cariotipo, en el estudio citogenético el hallazgo del cromosoma PH. y el análisis de la fosfatasa alcalina granulocitica. Además de la utilización y realización correcta de la historia clínica acompañada con la exploración cuidadosa de los casos analizados.
Tratamiento

Actualmente el tratamiento preferido para la LMC es el mesilato de imatinib, un inhibidor de la tirosina quinasa que ha permitido pasar de esperanza de vida de cuatro años en 2001 a una cronificación de la enfermedad en el 90% de los casos. El imatinib, producido por Novartis con el nombre de Glivec o Gleevec, ha sido objeto de batallas judiciales y estudiado como ejemplo del alto precio de los medicamentos contra el cáncer.
El trasplante de médula ósea (alogénico) es curativo, pero solo se emplea en la fase acelerada.
Pronóstico
Son factores de mal pronóstico la edad avanzada, muy marcada leucocitosis o proporción de blastos, esplenomegalia gigante, la afectación grave de las otras series (trombocitosis y anemia) y las alteraciones citogenéticas añadidas.
Véase también
- Leucemia mieloide aguda
- Leucopenia
- Leucemia
- Pancitopenia
- Síndrome hipereosinofílico
- Tumores infantiles
Bibliografía
- Harrison, T. R. et al (2006). «Oncología y hematología». Principios de Medicina Interna. Chile: McGraw-Hill Interamericana. ISBN 970-10-5166-1.
- Robbins, S. L. et al (2005). «Enfermedades de los leucocitos, los ganglios linfáticos, el bazo y el timo». Patología Estructural y Funcional. España: Elsevier España S.A. ISBN 978-84-8174-841-3.
Enlaces externos
- Leucemia Mieloide Crónica Leucemia Mieloide Crónica (CMLeukemia)
- Medline Plus Leucemia Mieloide Crónica
- Universidad de Navarra, Clínica Universitaria Leucemia Mieloide Crónica
- Síntomas de la Leucemia Mielode Crónica
- CáncerInfo España Leucemia Mieloide Crónica
- Asociación Española Contra el Cáncer
- The Leukemia and Lymphoma Society ¿Qué es la leucemia mielógena crónica?
- Noticias
| Control de autoridades |
|
|---|
-
 Datos: Q729735
Datos: Q729735
-
 Multimedia: Chronic myeloid leukemia / Q729735
Multimedia: Chronic myeloid leukemia / Q729735