
| Fibrosis Pulmonar Idiopática | ||
|---|---|---|
 Imagen por tomografía computarizada de tórax en alta resolución mostrando un alto grado de fibrosis en parénquima pulmonar.
| ||
| Especialidad | neumología | |
| Sinónimos | ||
| Alveolitis Fibrosante Criptogénica | ||
La fibrosis pulmonar idiopática (FPI) es una enfermedad crónica que se caracteriza por una disminución progresiva de la función pulmonar. El término fibrosis pulmonar hace referencia a la cicatrización del tejido pulmonar y es la causa del agravamiento de la disnea (falta de aliento). La fibrosis normalmente se asocia con un mal pronóstico. El término «idiopático» se utiliza debido a que aún se desconoce la causa de la fibrosis pulmonar.
Normalmente la FPI afecta a adultos de entre 50 y 70 años, en particular a aquellos con antecedentes de tabaquismo, y en mayor número a los hombres que a las mujeres.
La FPI pertenece a un gran grupo de más de 200 enfermedades pulmonares llamadas neumopatías intersticiales (NI) que se caracterizan por afectar principalmente al intersticio pulmonar, el tejido entre los alveolos pulmonares. Sin embargo estas enfermedades con frecuencia no solo afectan al intersticio, sino también al espacio alveolar, las vías respiratorias periféricas y a los vasos sanguíneos. El tejido pulmonar de los afectados por la FPI muestra un patrón histopatológico característico conocido como neumonía intersticial usual (NIU). Por lo tanto, la NIU es la contrapartida patológica de la FPI.
En 2011 se publicaron unas nuevas directrices para el diagnóstico y tratamiento de la FPI. El diagnóstico de FPI requiere la exclusión de otras causas habituales de NI y la presencia de un patrón radiológico típico que se identifica mediante tomografía computarizada de alta resolución (TCAR). En un entorno clínico adecuado es posible realizar el diagnóstico de la FPI basándose únicamente en la TCAR, evitándose así la necesidad de realizar una biopsia pulmonar quirúrgica.
Clasificación
La FPI pertenece a un gran grupo de más de 200 enfermedades pulmonares llamadas neumopatías intersticiales (NI) que se caracterizan por afectar al intersticio pulmonar, el tejido entre los alveolos pulmonares.La FPI es una enfermedad diferenciada entre las neumonías intersticiales idiopáticas (NII), las cuales a su vez son un tipo de NI, también conocidas como neumopatías parenquimatosas difusas (NPD).
La clasificación de 2002 de la American Thoracic Society/European Respiratory Society (ATS/ERS) para las NII se actualizó en 2013. En esta nueva clasificación existen tres categorías principales de NII: NII principal, NII raras y NII inclasificables. Las NII principales se agrupan en NI fibrosantes crónicas, que incluye la FPI y la neumonía intersticial no específica [NINE]), las NI relacionadas con el tabaquismo (por ejemplo, la neumopatía intersticial-bronquiolitis [NI-B] y la neumonía intersticial descamativa [NID]; y las NI agudas/subagudas (por ejemplo, neumonía organizante criptogénica [NOC] y neumonía intersticial aguda [NIA].
El diagnóstico de las NII requiere la exclusión de causas conocidas asociadas a las NI. Se pueden citar como ejemplo de NI de origen conocido la neumonitis por hipersensibilidad, histiocitosis pulmonar de la célula de Langerhan, asbestosis, aspiración crónica, y vasculopatías del colágeno. Sin embargo, estas enfermedades con frecuencia no solo afectan al intersticio, sino también al espacio alveolar, las vías respiratorias periféricas y los vasos sanguíneos.
La siguiente imagen muestra la nueva clasificación de las NII

Epidemiología
Aunque la FPI es una enfermedad rara, es la forma más común de todas las NII. La prevalencia de la FPI se ha estimado entre 14,0 y 42,7 por cada 100.000 personas, sobre la base de un análisis de datos de reclamaciones sanitarias en los EE. UU. (la variación en los valores es debida a diferencias en las definiciones de los casos empleados en los análisis). La FPI es mucho más común en hombres que en mujeres y normalmente se diagnostica en personas mayores de 50 años.
La incidencia de la FPI resulta difícil de determinar ya que no se han aplicado, de manera constante, unos criterios diagnósticos uniformes. Un estudio reciente en los Estados Unidos estimó una incidencia de 6,8-16,3 afectados por cada 100.000 habitantes. En los 27 países de la Unión Europea, diversas fuentes estiman una incidencia de 4,6–7,4 afectados por cada 100.000 habitantes, lo que sugiere que unos 30.000–35.000 nuevos pacientes son diagnosticados con FPI cada año.
Un reciente estudio observacional, retrospectivo de cohortes, en un único centro en el que se incluyeron pacientes con diagnóstico de NI en el Hospital Universitario de Aarhus (Dinamarca) entre 2003 y 2009 reveló una incidencia de 4,1 por 100.000 habitantes/año para la NI. La FPI fue el diagnóstico más común (28 %), seguida de la NI relacionada con conectivopatías (14 %), neumonitis por hipersensibilidad (7 %) y neumonía intersticial no específica (NINE) (7 %). La incidencia de la FPI fue de 1,3 por 100.000 habitantes/año.
Debido a la heterogénea distribución de la afección por los diferentes países europeos, deben actualizarse los datos epidemiológicos mediante un registro pan europeo para la NI y la FPI.
Causas y factores de riesgo de la FPI
Como su propio nombre indica la FPI o fibrosis pulmonar idiopática es idiopática, lo que significa que no se conoce su causa, pero ciertos factores medioambientales y las exposiciones han demostrado aumentar el riesgo de padecer la FPI. Se reconoce y acepta que el consumo de tabaco es el factor de riesgo más importante para la FPI, riesgo que se ve incrementado al doble. Se ha demostrado que otras exposiciones medioambientales y laborales, como la exposición al polvo de los metales, de la madera, del carbón, el silicio y el polvillo de talla, así como las ocupaciones relacionadas con la agricultura y ganadería, aumentan el riesgo de FPI. Existen ciertos indicios de que ciertas infecciones víricas pueden estar asociadas con la fibrosis pulmonar idiopática y otras neumopatías fibróticas.
Etiología y patología
A pesar de exhaustivas investigaciones, se continúa sin saber cuáles son las causas exactas de la FPI. La fibrosis presente en la FPI se ha vinculado al tabaquismo, factores medioambientales (p. ej. a la exposición a gases, humos, productos químicos o polvos en el entorno laboral), otras afecciones entre las que se incluyen la enfermedad por reflujo gastroesofágico (ERGE), o una predisposición genética (FPI familiar). Sin embargo, ninguna de éstas se encuentra en todas las personas afectadas por la FPI y, por ello, no ofrecen una explicación completamente satisfactoria de la enfermedad.
Se cree que la FPI es el resultado de un proceso de cicatrización aberrante que incluye o afecta a un depósito anómalo y excesivo de colágeno (fibrosis) en el intersticio pulmonar con una inflamación mínima asociada.
La hipótesis actual es que la lesión inicial o repetida que ocurre en la FPI afecta a las células pulmonares llamadas epiteliocitos alveolares (EA), que recubren la mayoría de la superficie alveolar. Cuando se dañan o pierden EA de tipo I, se cree que EA de tipo II inician una proliferación para cubrir las membranas basales que han quedado expuestas. En condiciones de reparación normal, el EA de tipo II hiperplástico muere y las células restantes se extienden y experimentan un proceso de diferenciación para convertirse en EA de tipo I. En procesos patológicos y en presencia del factor de crecimiento transformante beta (TGFβ), los fibroblastos se acumulan en estas zonas de daño y se diferencian en miofibroblastos que segregan colágeno y otras proteínas. Anteriormente se pensaba que la inflamación era el proceso inicial de la cicatrización del tejido pulmonar. Sin embargo, de acuerdo con los hallazgos más recientes, el desarrollo de los focos fibroblásticos antecede a la acumulación de células inflamatorias y el posterior depósito de colágeno.
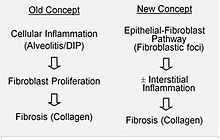
Este modelo patogénico lo apoyan, indirectamente, las características clínicas de la FPI, incluyendo un comienzo insidioso, una evolución a lo largo de varios años, reagudizaciones relativamente infrecuentes y una respuesta negativa al tratamiento con inmunosupresión. En la actualidad se encuentran en fase inicial de prueba, o en consideración para su futuro desarrollo, una serie de terapias cuyo objetivo es la activación del fibroblasto o la síntesis de matriz extracelular. La FPI familiar supone menos del 5 % del total de pacientes con FPI y es clínica e histológicamente indistinguible de la FPI esporádica. Las asociaciones genéticas incluyen mutaciones en las proteínas pulmonares tensioactivas A1, A2, C (SFTPA1, SFTPA2B) y la mucina (MUC5B). Un aspecto notable de la variante MUC5B es su elevada frecuencia de detección, ya que se encuentra en aproximadamente el 20 % de los individuos con ascendencia de Europa del norte y occidental, y en el 19 % de la población del Estudio del Corazón de Framingham. Las mutaciones en los genes de la telomerasa humana se asocian también con la fibrosis pulmonar familiar y, en algunos pacientes, con la FPI esporádica (TERT, TERC). Recientemente, se ha descrito una mutación vinculada al cromosoma X en un tercer gen asociado con la telomerasa, la disquerina (DKC1) en una familia con FPI.
Diagnóstico
El diagnóstico temprano de la FPI es un requisito previo para su tratamiento precoz, así como para una posible mejoría de los resultados asistenciales a lo largo de esta enfermedad progresiva y, al final, mortal. Si existe sospecha diagnóstica de FPI, su confirmación puede resultar difícil. Sin embargo, se ha demostrado que un enfoque multidisciplinar en el que participen un neumólogo, un radiólogo, un patólogo y un fisioterapeuta respiratorio, todos expertos en neumopatías intersticiales, mejora la precisión del diagnóstico de FPI.
El Multidisciplinary Consensus Statement on the Idiopathic Interstitial Pneumonias publicado por la American Thoracic Society (ATS) y la European Respiratory Society (ERS) en 2000 propuso una serie de criterios específicos principales y secundarios para establecer el diagnóstico de FPI. Sin embargo, en 2011, la ATS y la ERS, junto con la Japanese Respiratory Society (JRS) y la Latin American Thoracic Association (ALAT) publicaron una serie de criterios simplificados y actualizados para el diagnóstico y tratamiento de la FPI. En la actualidad, un diagnóstico de FPI requiere:
- La exclusión de causas conocidas de NI, p.ej., exposiciones domésticas o laborales, trastornos del tejido conjuntivo, o exposición/toxicidad a fármacos
- La presencia de un patrón radiológico típico de NIU en la TCAR.
En el entorno clínico adecuado es posible realizar el diagnóstico de la FPI basándose únicamente en la TCAR, evitándose así la necesidad de una biopsia pulmonar quirúrgica.
Reconocer la FPI en la práctica médica puede resultar problemático ya que los síntomas a menudo parecen similares a los de enfermedades más comunes como el asma, la enfermedad pulmonar obstructiva crónica (EPOC) y la insuficiencia cardíaca congestiva (www.diagnoseipf.com. El principal problema al que se enfrentan los facultativos es determinar si colectivamente la anamnesis, los síntomas (o signos), la exploración radiológica, y las pruebas pulmonares funcionales sugieren la presencia de FPI o si los hallazgos se deben a otros procesos. Desde hace tiempo se sabe que los pacientes con NI relacionada con la exposición al amianto, fármacos (como los agentes quimioterápicos o nitrofurantoin), con artritis reumatoidea y esclerodermia/esclerosis sistémica pueden resultar difíciles de distinguir de aquellos afectados por FPI. Otras consideraciones en el diagnóstico diferencial incluyen la neumopatía intersticial relacionada con la enfermedad mixta del tejido conjuntivo, sarcoidosis avanzada, neumonitis crónica por hipersensibilidad, histiocitosis pulmonar de la célula de Langerhan y fibrosis inducida por radiación.
Características clínicas

En muchos pacientes, los síntomas están presentes durante bastante tiempo antes del diagnóstico.Entre las características clínicas más habituales de la FPI se incluyen:
- Edad superior a los 50 años
- Tos seca, improductiva en esfuerzo
- Disnea de esfuerzo progresiva (dificultad al respirar durante el ejercicio)
- Estertores inspiratorios secos y bibasilares en la auscultación “con sonido de velcro” (un sonido crepitante en los pulmones durante la inhalación, parecido a aquel que se produce al separar lentamente un trozo de velcro, que se aprecia con un estetoscopio).
- Dedos hipocráticos, una desfiguración de las puntas de los dedos de las manos o de los pies (véase la imagen)
- Resultados anómalos en las pruebas pulmonares funcionales, con indicios de obstrucción y alteración en el intercambio gaseoso.
Estas características se deben a la deficiencia crónica de oxígeno en sangre y pueden estar presentes en una amplia variedad de otros trastornos pulmonares, no siendo específicas de la FPI. Sin embargo, debe considerarse la presencia de FPI en todos aquellos pacientes con disnea crónica de esfuerzo inexplicable que cursen con tos, estertores inspiratorios bibasilares o dedos hipocráticos.
La valoración de los estertores “tipo velcro” en la auscultación pulmonar es una forma práctica de mejorar el diagnóstico precoz de la FPI.El médico puede reconocer fácilmente los sutiles estertores característicos de la FPI.
Si se hallan sutiles estertores bilaterales durante todo el periodo inspiratorio que persisten tras varias inspiraciones profundas, y si continúan presentes en diferentes momentos con varias semanas de diferencia en un sujeto con edad ≥60 años, debería sospecharse la presencia de FPI y considerarse la opción de realizar una TCAR del tórax, la cual es más sensible que una radiografía del tórax. Debido a que los estertores no son un signo específico de la FPI, se debe realizar un proceso diagnóstico más exhaustivo.
El reconocimiento de estos síntomas requiere una serie de investigaciones adicionales con objeto de diagnosticar la FPI.
Radiología
Las radiografías de tórax resultan útiles para el seguimiento sistemático de los pacientes con FPI. Desafortunadamente, las radiografías de tórax estándar no ofrecen un diagnóstico, pero pueden revelar una reducción del volumen pulmonar, normalmente con marcas intersticiales reticulares prominentes cerca de las bases pulmonares.

La evaluación radiológica mediante TCAR es un elemento esencial en la ruta diagnóstica de la FPI. La TCAR se lleva a cabo con una gammacámara tomográfica axial computarizada convencional sin la inyección de agentes de contraste. Los cortes de evaluación son muy finos (1–2 mm).
La TCAR típica del tórax en un paciente con FPI muestra los cambios fibróticos en ambos pulmones, con una predilección por las bases pulmonares y la periferia.De acuerdo con las directrices conjuntas ATS/ERS/JRS/ALAT de 2011, la TCAR es un componente esencial en la ruta de diagnóstico de la FPI, la cual puede identificar NIU por la presencia de:
- Opacidades reticulares, a menudo asociadas con bronquiectasia por tracción
- Panalización manifestada como espacios quísticos agrupados, normalmente con diámetros de entre 3 y 10 mm, pero a veces de mayor tamaño. Normalmente subpleurales y que se caracterizan por paredes bien definidas y dispuestas en, al menos dos líneas. Generalmente, una línea de quiste no es suficiente para definir la panalización
- Las opacidades con aspecto de vidrio deslustrado son comunes pero menos amplias que la reticulación
- Distribución predominantemente basal y periférica aunque, a menudo, con patrón parcheado.

Histología
De acuerdo con las directrices actualizadas en 2011, ante la ausencia de un patrón típico de NIU en la TCAR, se requiere una biopsia pulmonar quirúrgica específica para confirmar el diagnóstico.
Las muestras histológicas para el diagnóstico de la FPI deben tomarse al menos de tres zonas diferentes y de un tamaño suficiente para que el patólogo pueda opinar sobre la arquitectura pulmonar subyacente. Las biopsias pequeñas, tales como las obtenidas mediante biopsia pulmonar transbronquial (realizadas durante una broncoscopia), normalmente no tienen el tamaño suficiente para lograr este propósito. Por ello, normalmente se necesitan biopsias mayores, obtenidas quirúrgicamente, mediante toracotomía o toracoscopia.
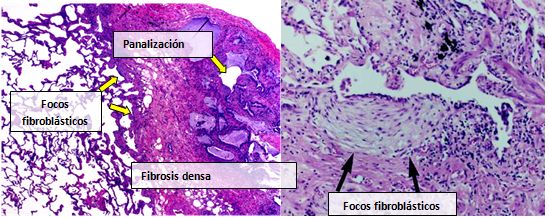
Normalmente, el tejido pulmonar de las personas afectadas por FPI muestra un patrón histopatológico de NIU característico y, por ello, es la contrapartida patológica de la FPI. Aunque a menudo un diagnóstico patológico de NIU se corresponde con un diagnóstico clínico de FPI, en otras enfermedades y fibrosis de origen conocido (enfermedades reumáticas, por ejemplo), también puede apreciarse un patrón histológico de NIU. Existen cuatro características clave de la NIU, incluida la fibrosis intersticial en un “patrón parcheado”, la cicatrización intersticial, los cambios por panalización y los focos de fibroblastos.
Los focos de fibroblastos son acumulaciones densas de miofibroblastos y tejido cicatricial, que, junto con la panalización, son los hallazgos patológicos principales que permiten un diagnóstico de NIU.
Lavado broncoalveolar
El lavado broncoalveolar (LBA) es un procedimiento diagnóstico bien tolerado en NI. Los análisis citológicos del LBA (fórmulas leucocíticas) deben tenerse en cuenta en la evaluación de los pacientes con FPI a criterio del especialista al cargo y basándose en la disponibilidad y experiencia en su centro asistencial. El LBA puede revelar diagnósticos específicos alternativos:cáncer, infecciones, neumonía eosinofílica, histiocitosis X, o proteinosisalveolar. En la evaluación de los pacientes en los cuales se sospecha FPI, la aplicación más importante del LBA es la exclusión de otros diagnósticos. Una linfocitosis preponderante (>30%) generalmente permite excluir el diagnóstico de FPI.
Pruebas pulmonares funcionales
Normalmente la espirometría revela una reducción de la capacidad vital (CV) con una reducción proporcional de las vías aéreas o un aumento de las vías aéreas para la capacidad vital observada. Este último caso refleja el aumento de la rigidez pulmonar (distensión pulmonar reducida) que se asocia con la fibrosis pulmonar, lo cual deriva en una mayor retracción elástica del pulmón.
La medición de los volúmenes pulmonares estáticos mediante pletismografía corporal u otras técnicas normalmente revela unos volúmenes pulmonares reducidos (restricción).Este hallazgo refleja la dificultad en inflar los pulmones fibróticos.
La capacidad de difusión del monóxido de carbono (DLCO) es invariablemente menor en pacientes con FPI y puede ser la única anomalía en los estadios leves o iniciales de la enfermedad. Su alteración resalta la propensión de los pacientes con FPI a mostrar una desaturación de oxígeno en esfuerzo, lo que puede evaluarse también con la prueba de marcha de 6 minutos(6MWT).
A veces se emplean términos como “leve”, “moderado” y “grave” para clasificar una enfermedad por grados y normalmente se basan en mediciones de pruebas pulmonares funcionales en reposo. Sin embargo, no existe un consenso claro respecto a la clasificación de los pacientes con FPI, ni cuáles son los mejores criterios y valores que deben utilizarse. La FPI leve o moderada se caracteriza por los siguientes criterios funcionales:
- Capacidad vital forzada (FVC) ≥50 %
- DLCO ≥30%
- Distancia en 6MWT ≥150 metros.
Asesoramiento genético en la FPI familiar
Un porcentaje estimado de entre el 10 y el 15 % de los pacientes con FPI presentan una forma de la enfermedad que afecta a familias, llamada fibrosis pulmonar familiar. Estudios recientes han identificado mutaciones genéticas asociadas con la fibrosis pulmonar familiar (véase información anterior) ,y en estos momentos, hay a disposición del público diversas pruebas para identificar dichas mutaciones.
El asesoramiento genético es el proceso por el que se facilita información a los pacientes y sus familias sobre la naturaleza, carácter hereditario e implicaciones de las enfermedades genéticas, con el objetivo de ayudarles a tomar decisiones, médicas y personales, con conocimiento de causa, así como para determinar el riesgo de padecer una enfermedad hereditaria. En aquellos casos en los que más de un miembro de la familia se encuentre afectado por fibrosis pulmonar, se deberá proporcionar asesoramiento genético, así como una identificación sistemática de mutaciones conocidas. El asesoramiento genético tras la realización de las pruebas de identificación de mutaciones puede ofrecer una interpretación personalizada de los resultados, un entendimiento del significado de los resultados para la salud del paciente, así como el impacto de éstos sobre el resto de los miembros de la familia.
Pronóstico

La evolución clínica de la FPI puede ser impredecible. El avance de la FPI se asocia con un periodo de supervivencia medio estimado de entre 2 y 5 años tras su diagnóstico. La supervivencia a 5 años para la FPI se estima entre el 20 y el 40 %, un índice de mortalidad superior al de varias neoplasias malignas, incluido el cáncer colorrectal, mieloma múltiple y el cáncer de vejiga.
Recientemente se ha propuesto un sistema de indexación y clasificación multidimensional para predecir la mortalidad por FPI. El nombre del índice es GAP y se basa en las iniciales en inglés de gender (sexo) [G], age (edad) [A], y dos variables pulmonares fisiológicas [P] (FVC y DLCO] que habitualmente se miden en la práctica médica para predecir la mortalidad de la FPI. El grado más alto del GAP (Grado III) se ha asociado con un 39 % de riesgo de mortalidad en 1 año. Este modelo también se ha evaluado en la FPI y otras NI, habiéndose mostrado unos buenos resultados en la predicción de la mortalidad en todos los subtipos principales de NI. Se ha desarrollado un índice NI-GAP modificado para su aplicación a los diferentes subtipos de NI con objeto de ofrecer estimaciones de supervivencia específicas de la enfermedad. En los pacientes de NI, la tasa de mortalidad general a 5 años es elevada, pero la tasa anual de mortalidad por cualquier causa en pacientes con alteración pulmonar leve o moderada, es relativamente baja. Este es el motivo por el que normalmente, en los estudios clínicos anuales de tratamientos para la FPI, se mide el cambio en la función pulmonar (FVC), en lugar de la supervivencia.
Además de los parámetros clínicos y fisiológicos para predecir la posible rapidez en evolución de los pacientes con FPI, las características genéticas y moleculares se asocian también con la mortalidad por FPI. Por ejemplo, se ha demostrado que los pacientes de FPI con un genotipo específico en el polimorfismo del gen MUC5B de la mucina (véase información anterior) experimentan un declive más lento de la FVC, así como una tasa de supervivencia notablemente mayor. Aunque dichos datos resultan interesantes desde el punto de vista científico, todavía resulta imposible aplicar un modelo pronóstico basado en genotipos específicos en la práctica médica rutinaria.
Tratamiento
Los objetivos del tratamiento de la FPI son, en esencia, reducir los síntomas, detener el avance de la enfermedad, evitar las reagudizaciones y prolongar la supervivencia. La atención preventiva, como por ejemplo las vacunas, y los tratamientos basados en los síntomas deben iniciarse en una etapa precoz en todos los pacientes.
Intervenciones farmacológicas
Se han investigado una serie de tratamientos para la FPI, incluidos los tratamientos con interferón gamma-1β,bosentan, ambrisentan, y anticoagulantes, pero en la actualidad ya no se consideran opciones de tratamiento eficaces.Muchos de los estudios anteriores se basaban en la hipótesis de que la FPI es un trastorno inflamatorio.
Pirfenidona
La pirfenidona es una pequeña molécula que combina efectos antiinflamatorios, antioxidantes y antifibróticos en modelos experimentales de fibrosis. La pirfenidona, comercializada bajo el nombre comercial de Esbriet®, está aprobada en Europa para el tratamiento de pacientes con FPI leve o moderada. También está aprobada para su uso en Japón, con nombre comercial Pirespa®, la India, China, Canadá, Argentina y México.
La pirfenidona se aprobó en la Unión Europea sobre la base de los resultados de tres estudios en fase III, aleatorizados, de doble ciego y controlado por placebo; uno de ellos llevado a cabo en Japón y los otros dos en Europa y los Estados Unidos (ensayos CAPACITY).
Un artículo de reseňa en la Cochrane Library (la revista de Cochrane Collaboration for evidence-based Medicine) basada en cuatro ensayos en los que participaron 1155 pacientes y que compara la pirfenidona con placebo, demostró una reducción significativa del 30 % en el riesgo de evolución de la enfermedad en los pacientes tratados con la pirfenidona. La FVC o CV también mejoró notablemente gracias a la pirfenidona, aunque una leve ralentización del declive de la FVC solo se pudo demonstrar en uno de los dos ensayos CAPACITY. Basándose en estos resultados mixtos, la American Federal Food and Drug Administration (FDA) exigió un tercer estudio clínico de fase III, ASCEND (NCT01366209). Este estudio, finalizado en 2014 y publicado en línea en el New England Journal of Medicine, mostró que la pirfenidona reducía notablemente el empeoramiento de la función pulmonar y la evolución de la FPI. Los datos del estudio ASCEND se agruparon con los datos procedentes de los dos estudios CAPACITY en un análisis pre-específico qué mostró que la pirfenidona reducía el riesgo de muerte en casi un 50 % tras un año de tratamiento.29 Basándose en estos resultados, la pirfenidona ha obtenido la calificación de Breakthrough Therapy por parte de La FDA norteamericana, una calificación reservada a fármacos para el tratamiento de enfermedades graves o potencialmente mortales, y para los cuales los estudios clínicos preliminares demuestran ìuna mejora sustancial respecto a las terapias existentes en uno o más criterios de valoración clínicamente importantes.
La empresa que ha desarrollado la pirfenidona, InterMune Inc., ofrece un uso por motivos humanitarios de la pirfenidona a través de un Programa de acceso extendido (PAE) multicéntrico en los Estados Unidos durante el periodo previo a su aprobación.
N-acetilcisteína y terapia triple
La N-acetilcisteína (NAC) es un precursor del glutatión, un antioxidante.Se ha planteado la hipótesis de que el tratamiento con dosis elevadas de NAC puede reparar un desequilibrio oxidante – antioxidante que se produce en el tejido pulmonar de los pacientes con FPI.En el primer ensayo clínico de 180 pacientes (IFIGENIA), se demostró que el NAC redujo el declive de la CV y el DLCO durante los 12 meses de seguimiento cuando se utilizaba de forma conjunta con prednisona y azatioprina (terapia triple).
Más recientemente, en los Estados Unidos de América, los National Institutes of Health (NIH) han llevado a cabo un estudio aleatorizado y controlado (PANTHER-IPF) con el objetivo de evaluar la terapia triple y la monoterapia con NAC en pacientes con FPI. Este estudio halló que la combinación de prednisona, azatioprina y NAC aumentaba el riesgo de muerte y hospitalizaciones, y el NIH anunció en 2012 que el grupo de terapia triple del estudio PANTHER-IPF había sido cancelado antes de tiempo. Este estudio también evaluaba la terapia solo con NAC y los resultados se esperan para el 2014. El estudio concluyó que, «en comparación con el placebo, la acetilcisteína no ofrecía ninguna ventaja significativa con respecto a la conservación de la FVC en pacientes con fibrosis pulmonar idiopática y una alteración leve o moderada en la función pulmonar».
Este estudio evaluó también la NAC sola, publicándose recientemente los resultados de este grupo del estudio en el New England Journal of Medicine, que mostraron que la monoterapia con NAC tampoco presenta ninguna ventaja significativa en pacientes con FPI leve o moderada.
Nintedanib (anteriormente BIBF 1120)
Se han finalizado dos ensayos en fase III (INPULSIS-1 y INPULSIS-2) con el tratamiento en desarrollo Nintedanib.30 Nintedanib es un inhibidor triple de la angiocinasa de administración oral y en fase de investigación clínica cuyo objetivo es el receptor de las tirosinas cinasas que participan en la regulación de la angiogénesis: el receptor del factor de crecimiento del fibroblasto (FGFR), el receptor del factor de crecimiento derivado de los trombocitos (PDGFR) y el receptor del factor de crecimiento endotelial vascular (VEGFR), y que también participan en la patogenia de la fibrosis y la FPI. En ambos ensayos en fase III, el nintedanib redujo notablemente el empeoramientode la función pulmonar en aproximadamente un 50 % a lo largo de un año.
Con respecto a los criterios de valoración secundarios, solo se produjo un aumento significativo en el tiempo (aparición tardía) de la primera reagudización (véase información anterior) en el grupo de nintedanib en comparación con el grupo de placebo en el ensayo INPULSIS-2. En el ensayo INPULSIS-1 no se observó este aumento.
El nintedanib, al igual que la pirfenidona, también ha sido aceptada para su presentación ante la FDA norteamericana, otorgándosele la calificación de Priority Review (Revisión prioritaria).
Futuras opciones terapéuticas
Actualmente, se están investigando diversos agentes en ensayos clínicos en fase III para la FPI, incluido los anticuerpos monoclonales simtuzumab, tralokimab, lebrikizumab y el FG-3019, un antagonista del receptor del ácido lisofosfatídico (BMS-986020). También se está llevando a cabo un estudio en fase II de la STX-100, dirigida contra diversos factores de crecimiento y citocinas, implicados en la proliferación, activación, diferenciación o supervivencia inapropiada de los fibroblastos.
Se puede encontrar más información en [www.ClinicalTrials.gov], un registro y base de datos de resultados de estudios clínicos llevados a cabo en todo el mundo en participantes humanos, subvencionados por entidades públicas y privadas.
Intervenciones no farmacológicas
Trasplante de pulmón
El trasplante de pulmón puede resultar apropiado para aquellos pacientes físicamente aptos para soportar una intervención de trasplante importante. En los pacientes con FPI, el trasplante de pulmón ha demostrado reducir el riesgo de muerte en un 75 % en comparación con los pacientes que permanecieron en la lista de espera. Desde la introducción de la Puntuación de asignación de pulmón (LAS), que prioriza a los candidatos al trasplante en función de su probabilidad de supervivencia, la FPI se ha convertido en la indicación más habitual para el trasplante de pulmón en los EE. UU.
Los pacientes sintomáticos con FPI, menores de 65 años, y con un índice de masa corporal (IMC) ≤26 kg/m², deben derivarse para el trasplante de pulmón, pero no existen datos claros que establezcan exactamente cuándo se debe realizar. Aunque controvertidos, los datos más recientes sugieren que el trasplante bilateral de pulmón es mejor que el trasplante de pulmón simple en pacientes con FPI. Las tasas de supervivencia a los cinco años después de un trasplante de pulmón en pacientes con FPI se estiman entre el 50 y el 56 %.
Oxigenoterapia a largo plazo (LTOT)
En las directrices para la FPI de 2011, la oxigenoterapia, u oxígeno suplementario para uso doméstico, pasó a ser una recomendación firme de uso en aquellos pacientes con hipoxemia en reposo clínicamente importante. Aunque no se haya demostrado que la terapia con oxígeno mejora la supervivencia en la FPI, algunos datos indican una mejora en la capacidad en esfuerzo.
Rehabilitación pulmonar
La fatiga y la pérdida de masa muscular son problemas comunes y discapacitantes para los pacientes con FPI. La rehabilitación pulmonar puede paliar los síntomas manifiestos de la FPI y mejorar el estado funcional al estabilizar o invertir las características extrapulmonares de la enfermedad. El número de estudios publicados sobre el papel de la rehabilitación pulmonar en la fibrosis pulmonar idiopática es reducido, pero la mayoría de dichos estudios han encontrado mejoras a corto plazo significativas en la tolerancia funcional al ejercicio, la calidad de vida y la disnea de esfuerzo. Normalmente en los programas de rehabilitación se incluye formación sobre actividades físicas, modulación nutricional, terapia ocupacional, así como asesoramiento educativo y psicosocial.
En la última fase de la enfermedad, los pacientes con FPI tienden a abandonar la actividad física debido a una creciente disnea. Siempre que sea posible, se deberá intentar convencer al paciente que no abandone la actividad física.
Cuidados paliativos
Los cuidados paliativos se centran en reducir los síntomas y mejorar el bienestar de los pacientes, más que tratar la enfermedad en sí. Estos incluyen tratar el agravamiento de ciertos síntomas con el uso de opioides crónicos para la disnea y tos grave. Además, la terapia con oxígeno puede resultar útil para la paliación de la disnea en pacientes hipoxémicos.
Los cuidados paliativos incluyen también reducir el sufrimiento físico y emocional, así como proporcionar apoyo psicosocial para los pacientes y sus cuidadores. A medida que avanza la enfermedad, los pacientes pueden experimentar miedo, angustia y depresión, y por ello debe considerarse el asesoramiento y la asistencia psicológica. En un estudio reciente llevado a cabo en pacientes ambulatorios con NI, incluida la FPI, se vio que la Spuntuación de depresión y el estado funcional (evaluado mediante una prueba de esfuerzo), así como la función pulmonar, influyen en la gravedad de la disnea.
En ciertos casos de disnea particularmente grave, puede considerarse la administración de morfina. La morfina puede reducir la disnea, la angustia y la tos sin reducir notablemente la saturación de oxígeno.
Tratamiento y seguimiento
Con frecuencia, la FPI no se diagnostica correctamente hasta que los datos fisiológicos o de las imágenes de diagnóstico sugieren la presencia de una NI, lo cual resulta en un retraso en la obtención de la asistencia adecuada. Si tenemos en cuenta que la FPI es una enfermedad con una supervivencia media de tres años tras su diagnóstico, la derivación precoz a un centro con los medios asistenciales adecuados debería considerarse para cualquier paciente ante la sospecha o confirmación de NI. Teniendo en cuenta el complejo diagnóstico diferencial de la FPI, y con objeto de lograr un diagnóstico preciso, es de máxima importancia planificar una reunión entre neumólogos, radiólogos y patólogos con experiencia en el diagnóstico de las NI.
Tras el diagnóstico de FPI y la elección del tratamiento apropiado de acuerdo con los síntomas y el estadio de la enfermedad, debe realizarse un estrecho seguimiento de su evolución. Debido a la gran variabilidad en el curso de la enfermedad, y a la elevada incidencia de complicaciones, como por ejemplo el cáncer pulmonar (hasta el 25% de los pacientes con FPI lo padecen), es obligatorio llevar a cabo una evaluación sistemática cada 3 o 6 meses; incluida una espirometría (pletismografía corporal), pruebas de capacidad de difusión, radiografías torácicas, 6MWT, evaluación de la disnea, calidad de vida, así como requerimientos de oxígeno.
Además, el conocimiento cada vez mayor de las complicaciones y afecciones comunes concomitantes que, con frecuencia, se asocian a la FPI requiere una evaluación sistemática de comorbilidades. La mayoría simplemente reflejan enfermedades concurrentes asociadas a la edad, y los medicamentos con sus interacciones y efectos secundarios.
Reagudización
La reagudización de la FPI (AE-IPF) se define como un empeoramiento inexplicable o la aparición de disnea en menos de 30 días con nuevos infiltrados radiológicos en la anomalía de la TCAR, a menudo superimpuestos sobre un fondo concordante con patrón de NIU. La incidencia anual de la AE-IPF se encuentra entre el 10 y el 15 % de todos los pacientes. El pronóstico de la AE-IPF no es bueno, con una mortalidad que varía del 78 al 96%. Deben excluirse otras causas de AE-IPF, tales como la embolia pulmonar, insuficiencia cardíaca congestiva, neumotórax o infección. La infección pulmonar debe descartarse mediante una aspiración endotraqueal o un LBA.
Muchos pacientes que sufren un deterioro agudo requieren tratamiento en cuidados intensivos, en particular cuando la insuficiencia respiratoria se asocia con inestabilidad hemodinámica, comorbilidades importantes o hipoxemia grave. La mortalidad durante la hospitalización es elevada. La ventilación mecánica solo debe aplicarse tras una evaluación cuidadosa del pronóstico del paciente a largo plazo y, siempre que sea posible, los deseos del mismo. Sin embargo, las directrices actuales desaconsejan el uso de ventilación mecánica en pacientes con insuficiencia respiratoria secundaria a la FPI.
Algunos casos bien documentados de FPI
- Marlon Brando, actor de cine y teatro
- Evel Knievel, legendario especialista de cine
- Steve Gerber, cocreador del personaje del libro cómico satírico Howard the Duck
- James “Scotty” Doohan, actor en la serie de televisión y cine Star Trek
- José Luis Rodríguez "El Puma", cantante venezolano
En otras especies
La FPI se ha detectado en varias razas de perros y gatos, aunque en donde mejor se ha caracterizado es en los West Highland White Terriers. Los animales con esta condición muestran muchas de las mismas manifestaciones clínicas que los humanos, incluida la intolerancia progresiva al esfuerzo, un aumento de la frecuencia respiratoria, y consiguientes problemas respiratorios. En general, el pronóstico es malo.
Enlaces externos
-
 Wikimedia Commons alberga una categoría multimedia sobre Fibrosis pulmonar idiopática.
Wikimedia Commons alberga una categoría multimedia sobre Fibrosis pulmonar idiopática. - FPI en MedlinePlus
- Comunidad de fibrosis pulmonar idiopática (FPI)
- Fundación de la fibrosis pulmonar
- Comunidad AIR
- AIMIP, Asociación italiana de enfermedades intersticiales o raras del pulmón Archivado el 9 de agosto de 2013 en Wayback Machine.
- el registro europeo de la FPI (eurIPFreg) se ha convertido en la principal base de datos europea de datos longitudinales de pacientes con FPI, incluidos grupos de control de pacientes con otras neumopatías
- La actividad de la ILD CARE FOUNDATION se centra en potenciar el conocimiento, apoyar la investigación, contribuir a la prevención y ofrecer asesoramiento en materia de neumopatías intersticiales.
- www.diagnoseipf.com
- KnowIPFNow.com
- inIPF
- IPFtoday.com
- ipfcharter.org Archivado el 22 de octubre de 2014 en Wayback Machine.
| Control de autoridades |
|
|---|
-
 Datos: Q2290446
Datos: Q2290446
-
Multimedia: Idiopathic pulmonary fibrosis / Q2290446