
| Enfermedad de Stargardt | ||
|---|---|---|
| Especialidad | oftalmología | |
| Síntomas | Pérdida de la visión central, baja agudeza visual | |
| Sinónimos | ||
|
Síndrome de Stargardt Fundus flavimaculatus, Distrofia y degeneración macular de Stargardt | ||
La enfermedad de Stargardt, también conocida como distrofia macular juvenil, es una enfermedad ocular hereditaria que se caracteriza por una degeneración macular, siendo la mácula la zona central de la retina dotada de la máxima sensibilidad que permite la visión fina de los detalles. El inicio de los síntomas tiene lugar en la adolescencia, aunque existe algún caso que ha tenido manifestaciones en la primera década de la vida. El paciente refiere que poco a poco deja de ver sin darse cuenta y se manifiesta por perdida de agudeza visual progresiva.
Historia

La primera descripción fue realizada en el año 1909 por el oftalmólogo alemán Karl Stargardt. En 1965 Franceschetti le dio el nombre de fundus flavimaculatus. Actualmente pueden denominarse indistintamente enfermedad de Stargardt o fundus flavimaculatus.
En 1997, los investigadores lograron aislar el gen de la distrofia macular de Stargardt: el ABCA4. Este gen produce una proteína que desempeña un papel importante en el transporte de energía desde y hacia las células fotorreceptoras de la retina. Esta enfermedad debe su nombre al oftalmólogo alemán Karl Stargardt aunque al principio se llamaba solamente distrofia macular juvenil. Actualmente no existe un tratamiento efectivo para combatir la patología causada por este alelo hereditario, pero disponer del conocimiento de las bases genéticas de la enfermedad puede ayudar a desarrollar nuevas estrategias terapéuticas para corregirla.
Frecuencia
Se considera una enfermedad rara, se presenta un caso por cada 10 000 personas aproximadamente.
Herencia
Es una enfermedad de transmisión hereditaria según un patrón autosómico recesivo. Está provocada por una mutación en el gen ABCA4, que codifica una proteína que sólo se expresa en la retina y que es un transportador de membrana de las células fotorreceptoras. Se conocen más de 558 mutaciones diferentes que pueden originar el mal.
Síntomas
Los síntomas más comunes al comienzo de esta enfermedad, suele referirse el paciente a que las cosas desaparecen y vuelven a aparecer, que se ven puntos negros que están y no están. Esto sucede en los primeros estadios y suele ser aproximadamente cinco años antes de que exista algún dato que verifique esta enfermedad. La retinografía está limpia y el paciente refiere que no ve ni con la mayor corrección posible. Cursa en ocasiones visión borrosa, zonas ciegas en el campo visual que se llaman escotomas y dificultad para adaptarse a las penumbras (lugares con poca luz). Aquellas personas que padecen la distrofia macular de Stargardt son muy sensibles a la luz y se ven obligados a usar gafas oscuras (filtros naranjas) así como visera o gorra. Deben evitar la ingesta de vitamina A y tomar el sol durante todo el año; para ello se pondrán crema solar y evitarán estar expuestos innecesariamente al sol.
Fondo de ojo
En las primeras fases la disminución de la agudeza visual es clara, pero no se observa grandes alteraciones en el fondo de ojo. A medida que progresa la enfermedad, fragmentos ricos en lípidos se acumulan en la capa del epitelio pigmentario de la retina por debajo de la mácula, los cuales aparecen como manchas amarillentas. El epitelio pigmentario de la retina es una capa que se encuentra entre la retina y la coroides, que es la responsable de irrigar y nutrir a las células fotorreceptoras de la retina que son los conos y bastones. Estos fragmentos de lípidos se llaman lipofuscina. En los casos avanzado de la enfermedad, esta acumulación progresiva de lipofuscina provoca la atrofia de la mácula y del epitelio pigmentario de la retina.
Características clínicas
Existe una amplia variedad de hallazgos clínicos:
- Mácula bermellón por epitelio pigmentario retiniano muy pigmentado
- Lo más característico son manchas rojo-amarillentas (flecks) por acumulación de lipofucsina a nivel del epitelio pigmentario retinal (EPR) que varían en forma, tamaño y distribución
- Signos: La fóvea puede ser normal o tener un moteado inespecífico; la atrofia geográfica puede tener una configuración en ojo de buey; la lesión foveal ovalada tiene un aspecto de "baba de caracol" o "bronce golpeado" en algunos casos rodeada de puntos blanco-amarillo.
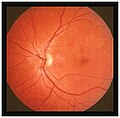
Estadios iniciales

Estadios iniciales

Estadio avanzado
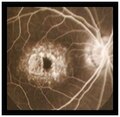
Fluoresceinografía digital

Microscopía electrónica de barrido





Tratamientos
Aún no existe tratamiento disponible para la enfermedad de Stargardt. Inclusive así, los siguientes tratamientos están en fase experimental:
- Terapia génica: aporte de una copia no mutante del gen afectado mediante un vector de transferencia génica viral portador del gen ABCA4 normal. (Stargen).
- Trasplante de células pigmentarias a partir de células madre embrionarias (Advanced Cell Technology).
- Trasplante de retina a partir de copas embrionarias generadas in vitro (Sasai).
- Fenretinide.
- Chips robóticos Argus II.
Las terapias actualmente en investigación sobre la enfermedad de Stargardt han sido revisadas recientemente.
Exámenes complementarios
- Campimetria que muestra la existencia de escotomas.
- Estudios eléctricos de retina: electrorretinografía y electrooculograma, pueden dar patrones variables incluso normales.
- Angiografía con fluoresceina: muestra una coroides oscura debido a depósitos de lipofuscina dentro del epitelio pigmentario de la retina e hiperfluorescencia macular debido al efecto ventana.
- Secuenciación genética para determinar la mutación de la que es portador el paciente. Es muy importante y es la prueba definitiva para poder saber cuál ha sido la mutación y en qué secuencia se ha producido. Ahora no es importante, pero lo será una vez que se desarrollen más ensayos clínicos con éxito. Es recomendable que todos los pacientes que tengan dudas o que su oftalmólogo les diga que esperen, que soliciten esta prueba, ya que con un análisis de sangre se puede determinar en aproximadamente seis meses.
Ayudas ópticas
Los pacientes sufren baja visión y aunque no se puede corregir con ningún tipo de anteojo, los avances en la fabricación digital de lentes permiten a día de hoy crear cristales con potencia suficiente para que combinándolos con ayudas ópticas les permitan realizar las actividades diarias con mayor fluidez.
- Anteojos para visión subnormal.
- Dispositivos electrónicos para baja visión
- Magnificadores, videolupas.
- Programas especiales para PC, programas lectores.
- Telescopios y lupas de mano.
- Apps para móvil; linterna, VoiceOver, etc.
Comunidad Stargardt en la UE
La Asociación D.O.C.E. (Discapacitados Otros Ciegos de España) propuso a RareConnect la organización de una comunidad internacional para poner en contacto a pacientes, amigos y simpatizantes de Stargardt con el fin de estar informados acerca de los últimos avances y posibles terapias cuando exista cura.
Véase también
- Stargardt APNES - [www.stargardt.com.ar]
- Stargardt Press - [1]
- D.O.C.E. - http://www.asociaciondoce.com
- [2]
| Control de autoridades |
|
|---|
-
 Datos: Q1317319
Datos: Q1317319
-
 Multimedia: Stargardt disease / Q1317319
Multimedia: Stargardt disease / Q1317319