
| Enfermedad de Rendu-Osler-Weber | ||
|---|---|---|
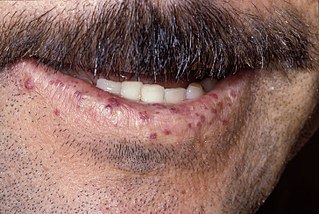 Telangiectasias características en labio inferior.
| ||
| Especialidad | genética médica | |
La telangiectasia hemorrágica hereditaria (HHT, del inglés Hereditary Hemorrhagic Telangiectasia), también conocida como Enfermedad de Osler-Weber-Rendu, es un trastorno genético que conduce a la formación anormal de los vasos sanguíneos en la piel, las mucosas, y a menudo en órganos tales como los pulmones, el hígado y el cerebro.
Todo ello puede conducir a hemorragias nasales, sangrado del tracto digestivo agudo y crónico, y diversos problemas debido a la afectación de otros órganos. El tratamiento se centra en la reducción del sangrado de las lesiones de los vasos sanguíneos, y a veces cirugía u otras intervenciones dirigidas a eliminar las malformaciones arteriovenosas (MAV) de órganos. El sangrado crónico a menudo requiere de suplementos de hierro y, a veces las transfusiones de sangre. La HHT se transmite de forma autosómica dominante, y se produce en uno de cada 5.000 personas.
La enfermedad lleva el nombre de Sir William Osler, Henri Jules Louis Marie Rendu y Frederick Parkes Weber, quienes la describieron a finales del siglo XIX y principios del siglo XX.
Signos y síntomas
Telangiectasias
Las telangiectasias pueden ocurrir en la piel y la mucosa revestimiento de la nariz y el tracto gastrointestinal. El problema más común es la epistaxis (sangrado nasal), que ocurre con frecuencia en la infancia y afecta a aproximadamente el 90-95% de las personas con HHT. Las lesiones en la piel y en la boca sangran con menos frecuencia, pero pueden considerarse cosméticamente desagradables y afectan a alrededor del 80%. Las lesiones cutáneas particularmente ocurren en los labios, la nariz y los dedos, y en la piel de la cara en áreas expuestas al sol. Aparecen de repente, y aumentan con el tiempo.
Alrededor del 20% se ven afectados por lesiones sintomáticas del tubo digestivo, aunque un porcentaje más alto tienen lesiones que no causan síntomas. Estas lesiones pueden sangrar intermitentemente, lo que rara vez es suficientemente importante como para ser notado (en forma de vómitos con sangre o heces de color negro), pero con el tiempo puede conducir a la disminución de hierro en el cuerpo, dando lugar a anemia por deficiencia de hierro.
Malformación arteriovenosa
Malformación arteriovenosa (MVA, malformaciones vasculares más grandes) se producen en los órganos más grandes, predominantemente los pulmones (50%), el hígado (30-70%) y el cerebro (10%), con una proporción muy pequeña (<1%) que tiene las MAV en la médula espinal.
Las malformaciones vasculares en los pulmones pueden causar una serie de problemas. Los pulmones normalmente "filtran" las bacterias y los coágulos de sangre de la circulación sanguínea; MAV eluden la red capilar de los pulmones y permitir que éstos migren hacia el cerebro, donde las bacterias pueden causar un absceso cerebral y coágulos de sangre pueden conducir a accidentes cerebrovasculares. HHT es la causa más común de malformaciones arteriovenosas pulmonares: entre todas las personas encuentran para tener malformaciones arteriovenosas pulmonares, el 70-80% se deben a HHT. Sangrado de las MAV pulmonares es relativamente poco común, pero puede causar hemoptisis (tos con sangre) o hemotórax (acumulación de sangre en la cavidad torácica). Las malformaciones vasculares grandes en el pulmón permiten sangre pobre en oxígeno desde el ventrículo derecho a pasar por alto los alvéolos, lo que significa que la sangre no tiene un oportunidad de absorber oxígeno fresco. Esto puede conducir a la falta de aire. Las grandes MAV pueden llevar a platipnea, dificultad en la respiración que es más marcada al sentarse frente al acostarse,.. Esto probablemente refleja los cambios en el flujo sanguíneo asociadas con la posición. Muy grande MAV causan una marcada incapacidad para absorber oxígeno, que puede ser observado por cianosis (coloración azulada de los labios y la piel), discotecas de las uñas de las manos (a menudo se encuentran en los niveles crónicamente bajos de oxígeno), y un zumbido sobre la parte afectada del pulmón detectables por el estetoscopio.
Los síntomas producidos por las MAV en el hígado dependen del tipo de conexión anormal que se forman entre los vasos sanguíneos. Si la conexión es entre las arterias y las venas, una gran cantidad de sangre que pasa por los órganos del cuerpo, para el cual el corazón compensa mediante el aumento del gasto cardíaco. Insuficiencia cardiaca congestiva Eventualmente desarrolla ("alta salida de insuficiencia cardiaca"), con falta de aliento y la hinchazón de las piernas, entre otros problemas. Si la MAV crea una conexión entre la vena porta y los vasos sanguíneos del hígado, el resultado puede ser la hipertensión portal (aumento de la presión de la vena porta), en la que se forman vasos sanguíneos colaterales en el esófago (varices esofágicas), que pueden sangrar violentamente, por otro lado, el aumento de la presión puede dar lugar a la acumulación de fluido en la cavidad abdominal (ascitis). Si el flujo dentro de la MAV se encuentra en la otra dirección, la sangre venosa portal fluye directamente en las venas en lugar de correr a través del hígado, lo que puede conducir a la encefalopatía hepática (confusión debido a los productos de desecho portal irritantes el cerebro). En raras ocasiones, los conductos biliares se ven privados de la sangre, dando lugar a colangitis severa (inflamación de los conductos biliares). MAVs hepáticos son detectables en más del 70% de las personas con HHT, pero solo el 10% tienen problemas como consecuencia.
En el cerebro, las MAV ocasionalmente ejercen presión, dando lugar a dolores de cabeza. Ellos también pueden aumentar el riesgo de convulsiones, como lo haría cualquier tejido anormal en el cerebro. Por último, la hemorragia de una MAV puede conducir a una hemorragia intracerebral (sangrado en el cerebro), lo que hace que cualquiera de los síntomas de un derrame cerebral, como debilidad en una parte del cuerpo o dificultad para hablar. Si el sangrado se produce en el espacio subaracnoideo (hemorragia subaracnoidea), por lo general hay una severa, dolor de cabeza repentino y el nivel de la conciencia y, a menudo debilidad disminuyó en parte del cuerpo.
Otros problemas
Una proporción muy pequeña (los afectados por mutaciones SMAD4, ver más abajo) tiene múltiples pólipos benignos en el intestino grueso, que pueden sangrar o transformar en cáncer colorrectal. Una proporción igualmente pequeña experimenta hipertensión pulmonar, un estado en el que se aumenta la presión en las arterias pulmonares, ejerciendo presión en el lado derecho del corazón y causando edema periférico (hinchazón de las piernas), desmayos y ataques de dolor en el pecho. Se ha observado que el riesgo de trombosis (especialmente la trombosis venosa, en la forma de la trombosis venosa profunda o embolia pulmonar) puede ser aumentada. Existe la sospecha de que las personas con HHT pueden tener una inmunodeficiencia leve y por tanto corren un riesgo ligeramente mayor de contraer infecciones.
Genética

La telangiectasia hemorrágica hereditaria tiene un patrón de herencia autosómico dominante.
HHT es un trastorno genético por definición. Se hereda de forma autosómica dominante, lo que significa que una persona afectada lleva un gen anormal, con un 50% de probabilidades de transmitir el gen a sus descendientes. Las personas con síntomas de HHT que no tienen familiares con la enfermedad pueden tener una nueva mutación. Parece que lleva dos copias anormales del gen no es compatible con la vida, y por lo tanto no se han descrito los homocigotos.
Se reconocen cinco tipos genéticos de HHT. De éstos, tres han sido relacionados con determinados genes, mientras que los dos restantes han sido actualmente sólo asociado a un determinado lugar. Más de 80% de todos los casos de HHT se deben a mutaciones en cualquiera ENG o ACVRL1. Se sabe que un total de más de 600 mutaciones diferentes. No es probable que sea un predominio de cualquiera de los tipos en poblaciones particulares, pero los datos son contradictorios. MADH4 mutaciones, que causan poliposis colónica además de HHT, comprenden de alrededor de 2% de las mutaciones causantes de enfermedades. Aparte de MADH4, no está claro si las mutaciones en ENG y ACVRL1 conducir a síntomas particulares, aunque algunos informes sugieren que las mutaciones ENG son más propensos a causar problemas en los pulmones, mientras que ACVRL1 mutaciones pueden causar más problemas en el hígado, y la hipertensión pulmonar puede ser un problema particular en las personas con mutaciones ACVRL1. Las personas con exactamente las mismas mutaciones pueden tener diferente naturaleza y la gravedad de los síntomas, lo que sugiere que los genes adicionales u otros factores de riesgo pueden determinar la velocidad a la que las lesiones se desarrollan; estos aún no han sido identificados.
Nombre OMIM Gen Locus Descripción HHT1 187300 ENG 9q34.1 códigos ENG para endoglina, un receptor de TGF-β1 (factor de crecimiento transformante beta 1) y TGF-β3, el ligamiento genético se identificó en 1994. Se ha observado una alta proporción de mutaciones de desplazamiento del marco. Prácticamente todas las mutaciones se producen en la parte extracelular de la proteína (la parte que se encuentra en la superficie de la célula).
HHT2 600.376 ACVRL1 12q11-q14 ACVRL1 códigos para Alk-1 (receptor de activina-quinasa 1), un receptor de TGF-β1; ligamiento genético fue identificado en 1996.
HHT3 601.101 Desconocido 5q31 función desconocida, varillaje identificado en 2005.
HHT4 610.655 Desconocida 7p14. Función desconocida, la vinculación identificado en 2006.
JPHT 175.050 MADH4 18q21.1 MADH4 códigos para SMAD4, una proteína de señalización intracelular de los receptores de la superfamilia TGF. Las mutaciones en este gen causa HHT y la poliposis juvenil. Vinculación fue identificado en 2004. Las mutaciones principalmente en los exones 8-11, a menudo de novo (recién adquirida, no hereditaria).
Fisiopatología
Una representación esquemática de la ruta de señalización de TGF-β. Endoglina (amarillo) es necesaria para la señalización. El ligando (azul) se une al receptor complejo; rojo indica una proteína del receptor de tipo II, que activa un receptor de la proteína I (turquesa), tales como alqu-1, que a su vez fosforila un factor de transcripción nuclear Smad-basado (tipo verde y púrpura).
Las telangiectasias y malformaciones arteriovenosas en HHT se cree que surgen debido a cambios en la angiogénesis, el desarrollo de vasos sanguíneos de los ya existentes. El desarrollo de un nuevos vasos sanguíneos requiere la activación y migración de varios tipos de células, principalmente endotelio, músculo liso y pericitos. El mecanismo exacto por el cual las mutaciones HHT influyen en este proceso aún no está claro, y es probable que interrumpen un equilibrio entre las señales pro-y anti-angiogénica en los vasos sanguíneos. La pared de las telangiectasias es inusualmente friable, lo que explica la tendencia de estas lesiones a sangrar.
Todos los genes que se sabe lo que va a estar relacionados con HHT codifican para las proteínas de la vía de señalización de TGF-β. Este es un grupo de proteínas que participa en la transducción de señales de las hormonas de la superfamilia del factor beta de crecimiento de transformación (el factor beta transformador del crecimiento, proteína morfogenética ósea y las clases de factores de diferenciación del crecimiento), específicamente BMP9/GDF2 y Bmp10. Las hormonas no entran en la célula, pero enlace a los receptores en la membrana de la célula, los cuales a continuación, activan otras proteínas, eventualmente influir en el comportamiento celular en un número de maneras, tales como la supervivencia celular, la proliferación (aumentando en número) y la diferenciación (cada vez más especializada). Para la señal de la hormona para ser adecuadamente transducidas, se necesita una combinación de proteínas: dos de cada uno de los dos tipos de serina / receptores de membrana de tipo quinasa treonina específicos y endoglina. Cuando se une a la hormona, el receptor de tipo II fosforila proteínas (transferencia de fosfato) a las proteínas del receptor de tipo I (de las cuales Alk-1 es uno), que a su vez fosforilan un complejo de proteínas Smad (principalmente Smad1, Smad5 y Smad8). Estos se unen a SMAD4 y migrar al núcleo celular donde actúan como factores de transcripción y participar en la transcripción de genes particulares. Además de la vía Smad, los receptores de membrana también actuar en la vía MAPK, que tiene acciones adicionales sobre el comportamiento de las células. Tanto Alk-1 y la endoglina se expresan predominantemente en el endotelio, quizás explicar por qué las mutaciones que causan en HHT estas proteínas conducen principalmente a problemas de los vasos sanguíneos. [2] [8] Tanto ENG y ACVRL1 mutaciones conducen predominantemente a la subproducción de las proteínas relacionadas, en lugar de misfunctioning de las proteínas.
Diagnóstico
Los exámenes de diagnóstico pueden ser realizadas por varias razones. En primer lugar, se necesitan algunos exámenes para confirmar o descartar el diagnóstico. En segundo lugar, algunos son necesarios para identificar las complicaciones potenciales.
Telangiectasias
Una lesión vascular en el tracto digestivo, siendo tratado con plasma de argón.
La piel y telangiectasias cavidad oral son visualmente identificable en el examen físico, y del mismo modo que las lesiones en la nariz pueden ser vistas en la endoscopia de la nasofaringe o en la laringoscopia. La gravedad de la hemorragia nasal se puede cuantificar objetivamente el uso de un cuestionario de la red como en el que conste el número de episodios de sangrado por la nariz y su duración.
Telangiectasias del tracto digestivo pueden ser identificados en esofagogastroduodenoscopia (endoscopia de la parte del esófago, el estómago y primera del intestino delgado). Sólo se suelen llevar a cabo este procedimiento si hay anemia que es más marcada de lo esperado por la gravedad de las hemorragias nasales, o si hay evidencia de sangrado grave (vómitos con sangre, heces de color negro). Si el número de lesiones observadas en la endoscopia es inesperadamente baja, el resto del intestino delgado puede ser examinado con cápsula endoscópica, en la que el paciente traga un dispositivo con forma de cápsula que contiene una cámara en miniatura que transmite imágenes del tracto digestivo a una digital portátil grabadora.
Las malformaciones arteriovenosas
Identificación de las MAV requiere tratamiento de imágenes médicas detalladas de los órganos más comúnmente afectados por estas lesiones. No todas las malformaciones arteriovenosas causan síntomas o están en riesgo de hacerlo, y por lo tanto hay un grado de variación entre los especialistas en cuanto a si se realizan las investigaciones, y por qué modalidad, a menudo, las decisiones sobre este tema se alcanzan en conjunto con el paciente.
MAV pulmonares se puede sospechar por la aparición anormal de los pulmones en una radiografía de tórax, o hipoxia (bajo nivel de oxígeno) en la oximetría de pulso o la determinación de gases en sangre arterial. Ecocardiografía de contraste burbuja (burbuja de eco) puede ser utilizado como una herramienta de detección para identificar las conexiones anormales entre las arterias y las venas pulmonares. Esta consiste en la inyección de suero salino agitado en una vena, seguido por imágenes del corazón basado en ultrasonidos. Normalmente, los pulmones eliminan pequeñas burbujas de aire de la circulación, y están por lo tanto, sólo se observan en la aurícula derecha y el ventrículo derecho. Si una MAV está presente, aparecen burbujas en la aurícula izquierda y el ventrículo izquierdo, por lo general 3-10 ciclos cardíacos después de que el lado derecho, lo que es más lento que en los defectos del corazón, en la que hay conexiones directas entre el lado derecho e izquierdo del corazón . Un mayor número de burbujas es más probable que indique la presencia de un AVM. Echo burbuja no es una herramienta de detección perfecta, ya que se puede perder MAV pequeñas y no identificar el sitio de la MAV. A menudo, la tomografía computarizada con contraste (angiografía TC) se utiliza para identificar las lesiones pulmonares; esta modalidad tiene una sensibilidad de más del 90%. Puede ser posible omitir la administración de contraste en los modernos escáneres CT [5] La ecocardiografía.. también se utiliza si existe una sospecha de hipertensión pulmonar o insuficiencia cardíaca de alto rendimiento debido a las lesiones hepáticas grandes, a veces seguido de cateterismo cardíaco para medir las presiones dentro de las diversas cámaras del corazón.
CT-exploración de las malformaciones vasculares en el hígado en un paciente con un patrón de telangiectasia hemorrágica hereditaria que causa la perfusión no homogénea.
MAV hígado se puede sospechar por las pruebas de función hepática anormales en la sangre, debido a que los síntomas de la insuficiencia cardiaca se desarrollan, o debido a la ictericia u otros síntomas de disfunción hepática. La prueba de detección inicial más fiable es la ecografía Doppler del hígado, lo que tiene una sensibilidad muy alta para la identificación de las lesiones vasculares en el hígado. Si es necesario, la TC con contraste puede usarse para caracterizar mejor las MAV. Es muy común encontrar nódulos incidentales en exploraciones del hígado, por lo general debido a la hiperplasia nodular focal (HNF), ya que estos son cien veces más común en HHT en comparación con la población general. FNH es considerado como inofensivo. En general, los marcadores tumorales y las modalidades de formación de imágenes adicionales se utilizan para distinguir entre los tumores FNH y malignos del hígado. La biopsia hepática se recomienda en personas con HHT ya que el riesgo de hemorragia de las malformaciones arteriovenosas hepáticas puede ser significativo. Las exploraciones del hígado pueden ser útiles si alguien es sospechoso de HHT, pero no cumple con los criterios (véase más adelante) a menos que el hígado Las lesiones pueden ser demostrados.
MAV cerebrales pueden ser detectados en la angiografía por tomografía computarizada (ATC o angio TC) o la angiografía por resonancia magnética (ARM),. CTA es mejor en mostrar los vasos sí mismos, y MRA ofrece más detalles sobre la relación entre una MAV y el tejido cerebral circundante. En general, se recomienda la RM se pueden encontrar diversos tipos de malformaciones vasculares:.. malformaciones arteriovenosas, micro-malformaciones arteriovenosas, telangiectasias y las fístulas arteriovenosas [7] Si la cirugía, la embolización u otro tratamiento se contempla (véase más adelante ), la angiografía cerebral puede ser necesario para obtener suficiente detalle de los vasos. Este procedimiento conlleva un pequeño riesgo de accidente cerebrovascular (0,5%) y por lo tanto se limita a las circunstancias específicas. [7] [14] recientes directrices profesionales recomiendan que todos los niños con sospecha de HHT o definitiva experimentan una RM cerebral temprano en la vida para identificar malformaciones arteriovenosas que pueden causar complicaciones mayores. Otros sugieren que la detección de malformaciones arteriovenosas cerebrales es probablemente innecesario en aquellos que no están experimentando síntomas neurológicos, porque la mayoría de las lesiones descubiertas en las exploraciones de cribado no requieren tratamiento, creando interrogantes indeseables.
Las pruebas genéticas
Las pruebas genéticas están disponibles para el ENG, ACVRL1 y MADH4 mutaciones. Las pruebas no siempre es necesario para el diagnóstico, debido a que los síntomas son suficientes para distinguir la enfermedad de otros diagnósticos. Hay situaciones en las que las pruebas pueden ser particularmente útiles. En primer lugar, los niños y adultos jóvenes con un padre con HHT definitivo pueden tener síntomas limitados, sin embargo, estar en riesgo de algunas de las complicaciones antes mencionadas, si la mutación es conocida en el padre afectado, la ausencia de esta mutación en el niño evitaría la necesidad para las pruebas de detección. Por otra parte, las pruebas genéticas pueden confirmar el diagnóstico en los pacientes con síntomas limitados que de otro modo habrían sido etiquetados como "posible HHT" (ver más abajo).
El diagnóstico genético en HHT es difícil, ya que se producen mutaciones en numerosos lugares diferentes en los genes relacionados, sin mutaciones particulares que son muy frecuentes (en oposición a, por ejemplo, la mutación F508 de la fibrosis quística). El análisis de secuencia de los genes que participan por lo tanto, es el enfoque más útil (sensibilidad 75%), seguido por pruebas adicionales para detectar grandes deleciones y duplicaciones adicional (10%). No todas las mutaciones en estos genes se han relacionado con la enfermedad.
Las mutaciones en el gen MADH4 se asocia generalmente con poliposis juvenil, y la detección de una mutación indicaría una necesidad para cribar los familiares del paciente y afectada por pólipos y tumores del intestino grueso.
Criterios
El diagnóstico de realizarse en función de la presencia de los cuatro criterios, conocidos como los "criterios de Curaçao" [15] Si se cumplen tres o cuatro, el paciente tiene "HHT definido", mientras que dos da "posible HHT":
- Epistaxis recurrentes espontáneas.
- Múltiples telangiectasias en ubicaciones típicas (véase más arriba).
- Probada visceral AVM (pulmón, hígado, cerebro, columna vertebral).
- Miembro de la familia de primer grado con HHT.
A pesar de la designación "posible", alguien con un MAV visceral y una historia familiar pero sin hemorragias nasales o telangiectasias es todavía extremadamente propensos a tener HHT, debido a que estos MAVs son muy poco frecuentes en la población general. Al mismo tiempo, el mismo no puede decirse de las hemorragias nasales y telangiectasias dispersos, los cuales ocurren en personas no padecen esta enfermedad, en ausencia de malformaciones arteriovenosas. Estado de diagnóstico de alguien puede cambiar en el curso de la vida, ya que los niños pequeños no pueden presentar todos los síntomas, a los 16 años, trece por ciento son todavía indeterminado, mientras que a los 60 años la gran mayoría (99%) tienen una clasificación diagnóstica definitiva. Los hijos de pacientes con HHT establecidas pueden por lo tanto ser etiquetados como "posible HHT", un 50% puede llegar a tener HHT en el curso de su vida.
Tratamiento
El tratamiento de la HHT es sintomático (se trata de los síntomas y no la enfermedad en sí), ya que no existe un tratamiento que detiene el desarrollo de telangiectasias y malformaciones arteriovenosas directamente. Además, algunos tratamientos se aplican para prevenir el desarrollo de complicaciones comunes hemorragias nasales crónicas y hemorragias del tracto digestivo puede tanto dar lugar a la anemia. Si el propio sangrado no se puede detener por completo, la anemia requiere tratamiento con suplementos de hierro. Los que no pueden tolerar los comprimidos de hierro o soluciones pueden requerir la administración de hierro sacarosa por vía intravenosa y transfusión de sangre si la anemia está causando síntomas severos que requieren una rápida mejora de la cuenta de sangre.
La mayoría de los tratamientos utilizados en HHT se han descrito en los adultos, y la experiencia en el tratamiento de los niños es más limitada. Las mujeres con HHT que quedan embarazadas tienen un mayor riesgo de complicaciones, y se observan de cerca, aunque el riesgo absoluto sigue siendo bajo (1%).
Las hemorragias nasales
Un sangrado por la nariz aguda puede tratarse con una variedad de medidas, tales como el embalaje de la cavidad nasal con hisopos absorbentes o geles. La eliminación de los paquetes después de la hemorragia puede conducir a la reapertura de los vasos frágiles, y por lo tanto se recomienda engrasar o embalaje atraumática. Algunos pacientes pueden desear aprender embalamos para tratar hemorragias nasales sin tener que recurrir a la ayuda médica.
Frecuentes hemorragias nasales pueden ser impedido, en parte, al mantener las ventanas de la nariz húmeda, y mediante la aplicación de solución salina, que contiene estrógeno cremas o ácido tranexámico, los cuales tienen pocos efectos secundarios y pueden tener un pequeño grado de beneficio. Un número de modalidades adicionales tiene. sido utilizada para prevenir el sangrado recurrente, si las medidas simples no tienen éxito. Las terapias médicas incluyen el ácido tranexámico oral y estrógeno, la evidencia de estos es relativamente limitada, y el estrógeno es mal tolerado por los hombres y, posiblemente, conlleva riesgos de cáncer y enfermedades del corazón en mujeres que han pasado la menopausia. Nasal coagulación y cauterización puede. reducir el sangrado de telangiectasias, y se recomienda antes de considerar la cirugía, a menudo, se necesitan varias sesiones. Puede ser posible para embolizar lesiones vasculares a través de la radiología intervencionista, lo que requiere que pasa un catéter a través de una arteria grande y la localización de la arteria maxilar bajo la guía de rayos X, seguida de la inyección en el recipiente de partículas que ocluyen los vasos sanguíneos. La ventaja del procedimiento tiende a ser de corta duración, y puede ser más apropiado en los episodios de hemorragia grave.
Si han fallado otras intervenciones, varias operaciones se han reportado para proporcionar beneficio. Uno de ellos es dermoplasty septal o procedimiento de Saunders, [17] en el que la piel se trasplanta en las ventanas de la nariz, y el otro es el procedimiento de Young, en el que las ventanas de la nariz están sellados completamente.
La piel y el tracto digestivo
Las lesiones cutáneas de HHT pueden ser deformantes, y pueden responder al tratamiento con pulso largo Nd:. YAG Las lesiones cutáneas en las yemas de los dedos a veces puede sangrar y causar dolor. Los injertos de piel es a veces necesaria para tratar este problema.
Con respecto a lesiones del tubo digestivo, hemorragia leve y anemia leve resultante se trata con suplementos de hierro, y se administra ningún tratamiento específico. Existen datos limitados sobre el tratamiento hormonal y el ácido tranexámico para reducir el sangrado y anemia. Anemia o episodios de sangrado grave son tratados por vía endoscópica con coagulación con plasma de argón (APC) o el tratamiento con láser de las lesiones identificadas, lo que puede reducir la necesidad de tratamiento de apoyo. Los beneficios esperados no son tales que los reiterados intentos de lesiones el tratamiento se recomiendan. [7] súbita, hemorragia severa es inusual, si se encuentra, otras causas (como una úlcera péptica) deben tenerse en cuenta, pero la embolización puede haber utilizado en tales casos.
MAV pulmonares
Las lesiones pulmonares, una vez identificados, se suelen tratar de prevenir los episodios de sangrado y lo más importante a la embolia cerebral. Esto se hace sobre todo en lesiones de los vasos sanguíneos de alimentación de 3 mm o más grandes, ya que estos son los más propensos a causar complicaciones a largo plazo si no se trata. La terapia actual más eficaz es la embolización con espirales metálicas desmontables. El procedimiento consiste en la punción de una vena grande (por lo general bajo anestesia general), seguido por el avance de un catéter a través del ventrículo derecho y en la arteria pulmonar, después de lo cual se inyecta contraste radiológico para visualizar las malformaciones arteriovenosas (angiografía pulmonar). Una vez que la lesión ha sido identificado, bobinas se despliegan que obstruyen el flujo de sangre y permitir que la lesión a retroceder. En manos expertas, el procedimiento tiende a ser muy eficaz y con efectos secundarios limitados, pero las lesiones pueden reaparecer y más intentos que sean necesarios. Exploraciones CTA se repiten para monitorizar la recurrencia. La escisión quirúrgica ha esencialmente sido ahora abandonados debido al éxito de la embolización.
Las personas con cualquiera de las malformaciones arteriovenosas pulmonares definitivas o un ecocardiograma de contraste anormal sin lesiones visibles se consideran en riesgo de embolia cerebral. Por lo tanto, se les aconseja evitar el buceo, en el que las pequeñas burbujas de aire que se pueden formar en el bloodsteam que pueden migrar al cerebro y causar un derrame cerebral. Del mismo modo, se recomienda la profilaxis antimicrobiana durante los procedimientos en los que las bacterias pueden entrar en el torrente sanguíneo, como el trabajo dental, y la evitación de las burbujas de aire durante la terapia intravenosa.
MAV hígado
Dado que las malformaciones arteriovenosas hepáticas generalmente causan insuficiencia cardíaca de alto rendimiento, se hace hincapié en el tratamiento de esta con diuréticos para reducir el volumen de sangre circulante, restricción de sal y la ingesta de líquidos y fármacos antiarrítmicos en caso de ritmo cardíaco irregular. Esto puede ser suficiente en el tratamiento de los síntomas de hinchazón y dificultad para respirar. Si este tratamiento no es eficaz o conduce a efectos secundarios o complicaciones, la única opción que queda es el trasplante de hígado. Esto se reserva para pacientes con síntomas graves, ya que conlleva una mortalidad de alrededor del 10%, sino que conduce a buenos resultados si tiene éxito. El punto exacto en el que transplantion hígado se va a ofrecer aún no está completamente establecido. El tratamiento de embolización se ha intentado, pero conduce a complicaciones graves en una proporción de los pacientes y se desanime.
Otras complicaciones relacionadas con el hígado (hipertensión portal, varices esofágicas, ascitis, encefalopatía hepática) se tratan con las mismas modalidades tal como se utiliza en la cirrosis, aunque el uso de derivación portosistémica intrahepática transyugular tratamiento no se recomienda debido a la falta de beneficio documentado.
MAV cerebrales
La decisión de tratar MAV cerebrales depende de los síntomas que causan (como convulsiones o dolores de cabeza). El riesgo de sangrado está en relación con episodios previos de hemorragia, y si en la CTA o angiografía por resonancia magnética de la AVM parece estar profundamente arraigado o tiene drenaje venoso profundo. Tamaño de la MAV y la presencia de aneurismas parece importar menos. En HHT, algunas lesiones (fístulas de alto flujo arteriovenoso) tienden a causar más problemas, y el tratamiento está garantizado. Otros MAV pueden retroceder en el tiempo sin intervención. Diversas modalidades están disponibles, en función de la ubicación de la MAV y su tamaño: Cirugía, el tratamiento basado en la radiación y la embolización. A veces, múltiples modalidades se utilizan en la misma lesión.
Cirugía (por craneotomía, cirugía cerebral abierta) se puede ofrecer sobre la base de los riesgos del tratamiento según lo determinado por la escala Spetzler-Martin (grado IV), esta puntuación es mayor en las lesiones más grandes que están cerca de estructuras cerebrales importantes y tienen drenaje venoso profundo . Las lesiones de alto grado (IV y V) tienen un riesgo inaceptablemente alto y la cirugía no se ofrece normalmente en esos casos. Radiocirugía (usando la terapia de radiación específica tal como por un bisturí de rayos gamma) puede ser utilizado si la lesión es pequeña, pero cerca de estructuras vitales. Por último, la embolización se puede usar en las lesiones pequeñas que tienen sólo un único vaso de alimentación.
Los tratamientos experimentales
Varios fármacos antiangiogénicos aprobados para otras enfermedades, como el cáncer, se han investigado en ensayos clínicos pequeños. El anticuerpo anti-VEGF bevacizumab, por ejemplo, se ha utilizado fuera de la etiqueta en varios estudios. En el estudio más grande llevó a cabo hasta el momento, la infusión de bevacizumab se asoció con una disminución en el gasto cardiaco y reducción de la duración y el número de episodios de epistaxis en pacientes tratados con HHT. La talidomida, otro fármaco anti-angiogénesis, fue también informado que tiene efectos beneficiosos en pacientes HHT. Se encontró que el tratamiento talidomida para inducir maduración de los vasos en un modelo experimental de ratón de HHT y para reducir la gravedad y la frecuencia de hemorragias nasales en la mayoría de un pequeño grupo de pacientes HHT. Los niveles de hemoglobina en la sangre de estos pacientes tratados aumentó como resultado de la reducción de la hemorragia y la estabilización mejorada vaso sanguíneo.
Epidemiología
Las Antillas Neerlandesas, donde HHT es más común que en cualquier parte del mundo, situado en la costa de Venezuela.
Los estudios de población de muchas zonas del mundo han demostrado que la HHT se produce más o menos al mismo ritmo en casi todas las poblaciones: en algún lugar alrededor de 1 en 5000. En algunas zonas, es mucho más común, por ejemplo, en la región francesa de Haut Jura la tasa es de 1:2351 - dos veces más común que en otras poblaciones. Esto se ha atribuido a un efecto fundador, en el que una población desciende de un pequeño número de antepasados tiene una alta tasa de un rasgo genético particular, porque uno de estos antepasados albergaba este rasgo. En Jura, esto ha sido demostrado ser el resultado de una mutación particular ACVRL1 (llamado c.1112dupG o c.1112_1113insG). La mayor tasa de HHT es 1:1331, informó en Bonaire y Curaçao, dos islas del Caribe pertenecientes a las Antillas Neerlandesas.
La mayoría de las personas con HHT tienen una esperanza de vida normal. Las lesiones de la piel y hemorragias nasales tienden a desarrollarse durante la infancia. MAV son probablemente presentes desde el nacimiento, pero no necesariamente causan ningún síntoma. Hemorragias nasales frecuentes son el síntoma más común y puede afectar significativamente la calidad de vida.
Historia
Varios médicos ingleses del siglo decimonoveno, empezando por Henry Gawen Sutton (1836-1891) y seguido por Benjamin individuo Babington (1794-1866) y John Wickham Legg (1843 a 1921), se describen los más comunes características de HHT, en particular las hemorragias nasales recurrentes y la naturaleza hereditaria de la enfermedad. El médico francés Henri Jules Louis Marie Rendu (1844-1902) observó la piel y lesiones de la mucosa, y se distingue la condición de la hemofilia. El origen canadiense Sir William Osler (1849-1919), luego en el Hospital Johns Hopkins y más tarde en la Universidad de Oxford, hizo contribuciones adicionales con un informe de 1901 en el que se describen las lesiones características en el tracto digestivo. El inglés médico Frederick Parkes Weber (1863-1962) informó además de la condición en 1907 con una serie de casos. El término "telangiectasia hemorrágica hereditaria" fue utilizado por primera vez por el médico estadounidense Frederic M. Hanes (1883 a 1946) en un artículo de 1909 en el estado.
El diagnóstico de HHT permaneció una clínica hasta que los defectos genéticos que causan HHT fueron identificados por un grupo de investigadores de la Duke University Medical Center, en 1994 y 1996 respectivamente. En 2000, el comité internacional de asesoramiento científico de la Fundación HHT Internacional publicó los criterios de Curaçao ahora ampliamente utilizados. En 2006, un grupo de expertos internacionales se reunieron en Canadá y formuló una guía basada en la evidencia, patrocinado por la HHT Foundation International.
- Krupp, Marcus A. (1979). «Telangiectasia Hemorrágica Hereditaria». Análisis Clínico y Tratamiento. Ciudad de México: El Manual Moderno. ISBN 968-426-049-0.
- Asociación de afectados por el síndrome de Rendu-Osler-Weber.
| Control de autoridades |
|
|---|
-
 Datos: Q776881
Datos: Q776881
-
 Multimedia: Hereditary hemorrhagic telangiectasia / Q776881
Multimedia: Hereditary hemorrhagic telangiectasia / Q776881