
| Enfermedad de Krabbe | ||
|---|---|---|
| ||
| Especialidad | trastorno metabólico | |
| Síntomas |
infancia : retraso en el desarrollo , irritabilidad , espasticidad , hipotonía , microcefalia , atrofia óptica Juvenil : debilidad muscular , pérdida de visión , regresión del desarrollo Adulto : parestesias ardientes en las extremidades, pérdida de la destreza manual , debilidad muscular , neuropatía sensorial , atrofia muscular |
|
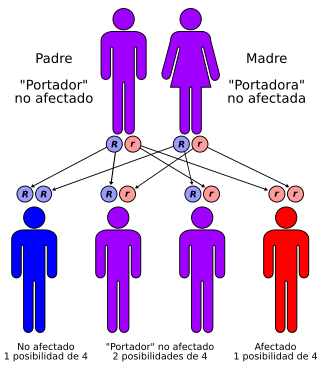
| Factores de riesgo | Padres heterocigotos (solo una copia) para la mutación del gen GALC | |
La enfermedad de Krabbe (también conocida como leucodistrofía globoide o lipidosis galactosilceramida) es una enfermedad autosómica recesiva que produce una acumulación de galactocerebrósidos debido a una deficiencia de la enzima galactocerebrosidasa. Esta acumulación de galactocerebrósidos provoca un trastorno en la vaina de mielina de la materia blanca del sistema nervioso (tanto central como periférico) que causan un trastorno neurológico degenerativo. Esta enfermedad recibe su nombre del neurólogo danés Knud Haraldsen Krabbe (1885-1965).
Incidencia
La enfermedad de Krabbe es una enfermedad rara que se da en aproximadamente 1 de cada 100 000 nacimientos[2], aunque se han registrado incidencias mayores en determinadas comunidades: en algunas comunidades árabes en Israel la incidencia es mucho mayor[3], 6 casos por cada 1000 nacimientos, y en los países escandinavos la incidencia es de 1 por cada 50 000 nacimientos.[4] La enfermedad de Krabbe también se ha encontrado en gatos [5] y perros.
Causas
Esta enfermedad está causada por una mutación en el gen que codifica la galactocerebrosidasa (GALC), localizado en el brazo pequeño del cromosoma 14 (14q31)[6], que produce una deficiencia en la enzima galactocerebrosidasa, lo que a su vez produce un aumento en la concentración de galactocerebrósidos. En casos aislados esta enfermedad puede estar provocada por una falta de la forma activa de la saposina A. La acumulación de lípidos no metabolizados afecta al crecimiento de la vaina protectora de mielina del nervio y provoca una gran degeneración de las habilidades motoras. En algunos casos también puede causar calcificaciones.[7] La enfermedad de Krabbe se clasifica dentro del grupo de trastornos conocido como leucodistrofías, enfermedades genéticas hereditarias que provocan una degeneración de la vaina de mielina que cubre las fibras nerviosas del cerebro.
La deficiencia de la enzima galactocerebrosidasa también produce un incremento de los niveles de otro glicosfingolípido, la Psicosina. Se ha propuesto que la Psicosina causa una degeneración de los axones en el sistema nervioso mediante la interrupción de las balsas lipídicas y puede desempeñar un papel en la enfermedad de Krabbe.[8][9]
Síntomas
Se han descrito dos variedades de la enfermedad de Krabbe. La de aparición temprana y la de aparición tardía, que muestran síntomas similares pero con algunas diferencias.
La más frecuente es la de aparición temprana, que se manifiesta en lactantes menores, normalmente entre los 3 y 6 meses de edad (aunque en algunos casos puede tardar unos años en manifestarse). Los síntomas incluyen irritabilidad, fiebres, rigidez en las extremidades, convulsiones, retraso en el desarrollo, dificultades en la alimentación, vómitos y disminución del desarrollo mental y motor. Estos síntomas son similares a los de la parálisis cerebral, por lo que en algunos diagnósticos se confunden ambas enfermedades. La enfermedad de Krabbe también puede producir los siguientes síntomas: debilidad muscular, espasticidad, sordera, atrofia óptica, sensibilidad a ruidos altos, ampliación del nervio óptico,[10] ceguera, parálisis y dificultad al tragar. También puede darse una pérdida de peso prolongada y nistagmo (movimiento involuntario e incontrolable de los ojos). Los niños afectados por esta variedad suelen morir a los pocos meses de comenzar a manifestarse los síntomas.
En cuanto a la variedad de aparición tardía, los síntomas son similares, aunque normalmente son más leve y su desarrollo es más lento. En este caso los primeros síntomas que suelen aparecer son los relacionados con la visión, seguidos de los problemas al caminar y de la rigidez muscular, aunque estos síntomas presentan grandes variaciones entre distintos paciente, incluso dentro de una misma familia.[7]
Detección
Existen varias pruebas que permiten detectar la enfermedad o sus síntomas: estudio de la retina del ojo para buscar el daño al nervio óptico, examen de sangre para medir los niveles de galactosilceramidasa en los glóbulos blancos, realizar una resonancia magnética de la cabeza, medir la velocidad de conducción nervioso o realizar pruebas moléculas para buscar mutaciones en el gen GALC.[11]
Diagnóstico molecular
Dado que no existe un tratamiento eficiente para la enfermedad conocido hasta la fecha, puede diagnosticarse en sujetos portadores con vistas a que recurran a técnicas de fecundación in vitro para evitar concebir hijos enfermos. Existen varios métodos para la detección del alelo mutado en portadores y no nacidos (diagnóstico prenatal):
- Ensayo enzimático: en la gran mayoría de los individuos enfermos se muestra una actividad anormalmente deficiente de la enzima galactosilceramidasa. Existen varios tipos de ensayos que pueden medir la actividad de esta enzima como la utilización de sustratos marcados radiactivamente. Sin embargo, este tipo de diagnóstico no es muy fiable en leucocitos y fibroblastos de la piel de sujetos portadores, ya que presentan una combinación enzimática defectuosa y sana que puede alterar el resultado del diagnóstico. Es el diagnóstico prenatal más fiable y económico en las clínicas en las que su uso está generalizado.
- Estudio genético. Se conoce una mutación en el gen galc que afecta a un 45 % de los enfermos europeos y a un 35 % de los enfermos de origen mexicano. Esta mutación consiste en una deleción de 30 kb en el gen galc y puede detectarse mediante estudios de mutegénesis inducida, secuenciación y estudios de delección/duplicación. Todos estos métodos son altamente fiables tanto en el rastreo de portadores como en el diagnóstico prenatal.
Tratamiento
Aunque a día de hoy no se conoce cura para esta enfermedad, sí que existen algunos tratamientos que pueden reducir sus síntomas.
Krivit et al. (1998)[12] realizó trasplantes alogénicos de células madre hematopoyéticas en niños con la enfermedad de Krabbe en los que ya produjeron anormalidades en el sistema nervioso central. Al realizar el tratamiento se comprobó que el deterioro del sistema nervioso central se revirtió y 14 meses después del trasplante no se produjo ninguna disminución esperada en la función del sistema nervioso central, por lo que se llegó a la conclusión de que estos trasplantes pueden revertir o prevenir las manifestaciones de la enfermedad de Krabbe en el sistema nervioso central.
Por otro lado, Escolar et al. (2005)[13] realizaron trasplantes de sangre del cordón umbilical de donantes no emparentados en 11 recién nacidos asintomáticos y 14 lactantes sintomáticos con la enfermedad de Krabbe infantil, a los que se les había administrado previamente quimioterapia ablativa. La proporción de trasplante de células donantes llevados con éxito y la supervivencia fueron del 100 % y 100 % (respectivamente) en los recién nacidos asintomáticos y del 100 % y 43 % (respectivamente) en el caso de los lactantes sintomáticos. Los pacientes que sobrevivieron mostraron unos niveles normales de galactocerebrosidasa en sangre. Los bebés asintomáticos mostraron un aumento de la mielinización progresivo y ganancias de habilidades de desarrollo. La mayoría de estos bebés presentaron unas funciones cognitivas y unas habilidades del lenguaje receptivo adecuadas a su edad, aunque algunos presentaron un retraso leve o moderado en el lenguaje expresivo y un retraso variable en la función motora. Los niños sometidos al trasplante después de la aparición de los síntomas presentaron una mejora neurológica mínima.
También se ha demostrado efectos beneficiosos del trasplante de médula ósea en el tratamiento de esta enfermedad, siempre y cuando se realice en casos tempranos. Del mismo modo, la fisioterapia puede ayudar a mantener y recuperar el tono muscular y la circulación.
2. "Krabbe disease". Genetics Home Reference. United States National Library of Medicine. 2008-05-02. Retrieved 2008-05-07.
3. Zlotogora J (Sep 1997). "Autosomal recessive diseases among palestinian Arabs". Journal of Medical Genetics 34(9): 765–766. doi:10.1136/jmg.34.9.765. ISSN 0022-2593. OCLC 120296280. PMC 1051064.PMID 9321766.
4. Books.Google.com
5. Salvadori C, Modenato M, Corlazzoli DS, Arispici M, Cantile C (May 2005). "Clinicopathological features of globoid cell leucodystrophy in cats". J. Comp. Pathol. 132 (4): 350–6.doi:10.1016/j.jcpa.2004.12.001. PMID 15893994.
6. "Regional mapping of the human galactocerebrosidase gene (GALC) to 14q31 by in situ hybridization".doi:10.1159/000133703.
7. Tappino, B., Biancheri, R., Mort, M., Regis, S., Corsolini, F., Rossi, A., Stroppiano, M., Lualdi, S., Fiumara, A., Bembi, B., Di Rocco, M., Cooper, D. N., Filocamo, M. "Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease." Hum. Mutat. 31: E1894-1914, 2010. Note: Electronic Article. [PubMed: 20886637, related citations] [Full Text: John Wiley & Sons, Inc.]
8. Lee, Wing; Kang, Dongcheul; Causevic, Ena; Herdt, Aimee; Eckman, Elizabeth; Eckman, Christopher (2010). "Molecular Characterization of Mutations That Cause Globoid Cell Leukodystrophy and Pharmacological Rescue Using Small Molecule Chemical Chaperones". The Journal of Neuroscience 30 (16): 5489–5497.doi:10.1523/JNEUROSCI.6383-09.2010.PMID 20410102.
9. White, Adam; Givogri, Maria; Lopez-Rosas, Aurora; Cao, Hongmei; Breemen, Richard van; Thinakaran, Gopal; Bongarzone, Ernesto (2009). "Psychosine Accumulates in Membrane Microdomains in the Brain of Krabbe Patients, Disrupting the Raft Architecture". The Journal of Neuroscience 29 (19): 6068–6077.doi:10.1523/JNEUROSCI.5597-08.2009.PMID 19439584.
10. Hussain, S. A.; Zimmerman, H. H.; Abdul-Rahman, O. A.; Hussaini, S. M.; Parker, C. C.; Khan, M. (May 2011). "Optic Nerve Enlargement in Krabbe Disease: A Pathophysiologic and Clinical Perspective". Journal of Child Neurology 26 (5): 642 644. doi:10.1177/0883073810387929.PMID 21285037. edit
11. Enfermedad de Krabbe en el sitio MedlinePlus
12. Krivit, W., Shapiro, E. G., Peters, C., Wagner, J. E., Cornu, G., Kurtzberg, J., Wenger, D. A., Kolodny, E. H., Vanier, M. T., Loes, D. J., Dusenbery, K., Lockman, L. A. "Hematopoietic stem-cell transplantation in globoid-cell leukodystrophy." New Eng. J. Med. 338: 1119-1126, 1998. [PubMed: 9545360, related citations] [Full Text: Atypon]
13. Escolar, M. L., Poe, M. D., Provenzale, J. M., Richards, K. C., Allison, J., Wood, S., Wenger, D. A., Pietryga, D., Wall, D., Champagne, M., Morse, R., Krivit, W., Kurtzberg, J. "Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease." New Eng. J. Med. 352: 2069-2081, 2005. [PubMed: 15901860, related citations] [Full Text: Atypon]
Enlaces externos
- Fundación Stennis
- sitio myspace Fundación Stennis
- Fundación Hunter Hope
- http://www.krabbes.com/trevors_trouble/scholarship.htm
- http://liamsmemorialfoundation.org/
| Control de autoridades |
|
|---|
-
 Datos: Q511372
Datos: Q511372
-
 Multimedia: Krabbe disease / Q511372
Multimedia: Krabbe disease / Q511372