
| Beta-talasemia | ||
|---|---|---|
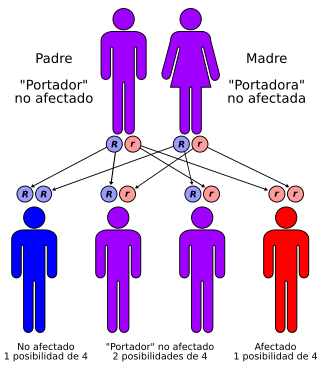 Beta-talasemia se transmite con un patrón fundamentalmente autosómico recesivo
| ||
| Especialidad | hematología | |
La beta-talasemia es una forma de talasemia caracterizada por un déficit en la síntesis de cadenas beta de la hemoglobina. La mayoría de los casos tiene su origen en una mutación del gen HBB en el cromosoma 11. También existen casos de deleciones de diversos tamaños que pueden afectar al gen de la beta globina o a la región de control del locus. Mayoritariamente es una enfermedad hereditaria con un patrón autosómico recesivo, pero también existen algunos casos donde la herencia es autosómica dominante. El resultado es que aumentan otros tipos de hemoglobina que no liberan el oxígeno con tanta facilidad, y los tejidos reciben menos oxigenación. Existen dos variedades de beta-talasemia (mayor o menor) según sea un déficit total o parcial de la síntesis. Aumenta la posibilidad de que se rompan los hematíes (hemólisis). Es una anemia que no se trata con hierro.
Causas moleculares de la enfermedad
La beta talasemia se puede producir por una disminución (beta-;β-) o una ausencia total (beta0; β0) de la síntesis de las cadenas de beta globina de la hemoglobina. Las cadenas beta de la globina están codificadas por el gen HBB, localizado en el brazo corto del cromosoma 11 (11p15.5) y contiene 3 exones y las regiones 5´y 3´ UTR. Se han descrito más de 200 mutaciones causantes de beta talasemia, la mayoría debidas a mutaciones de un solo nucleótido, pequeñas deleciones o inserciones que dan lugar a alteraciones del patrón de lectura (mutaciones de frameshift). Raramente debido a grandes deleciones. Teniendo en cuenta el defecto genético podemos clasificarlas como:
- β- talasemias Generalmente debidas a mutaciones que afectan a la región promotora del gen HBB. Producen una disminución de la síntesis de cadenas de beta globina dando lugar a fenotipos variables dependiendo de cuanto se reduce ésta.
- β0 talasemias Cursan con una ausencia completa de la síntesis de cadenas beta. Causadas principalmente por deleciones, mutaciones sin sentido y frameshift entre otros.
Diagnóstico molecular
Las mutaciones que afectan al gen HBB se detectan principalmente mediante la reacción en cadena de la polimerasa (PCR). Para ello se emplean primers o cebadores específicos de las mutaciones más frecuentes en la población originaria del paciente. Así por ejemplo la mutación β039, que introduce un codón de parada temprano, es la principal causa de β Talasemia (herencia autosómica recesiva) en la población de la isla de Cerdeña. Otras técnicas rápidas son la secuenciación directa, la PCR a tiempo real o en el caso de deleciones que afectan a regiones amplias del gen, mediante la técnica de MLPA.
Tipos de beta talasemia
La beta talasemia (anemia de Cooley)
La talasemia es un trastorno hereditario que afecta la producción de hemoglobina normal (un tipo de proteína presente en los glóbulos rojos cuya función es transportar oxígeno a los tejidos del cuerpo). La talasemia incluye varias formas diferentes de anemia. La gravedad y el tipo de anemia dependen del número de genes que estén afectados. La beta talasemia es causada por mutaciones en la cadena beta de la molécula de hemoglobina. Existe un gen para la cadena beta en cada cromosoma número 11, con un total de dos genes. La forma en que se alteran estos genes determina el tipo específico de beta talasemia en un niño:
-Beta talasemia grave (anemia de Cooley) - ambos genes de la cadena beta tienen deleciones, causando el tipo más grave de beta talasemia. Los pacientes que tienen talasemia grave necesitan frecuentes transfusiones de sangre y puede que no vivan mucho tiempo. Durante el primer año o dos primeros años de vida, pueden estar pálidos, irritables, tener poco apetito y padecer muchas infecciones. Sin tratamiento, aumenta el tamaño del hígado, del bazo y del corazón, y los huesos pueden volverse delgados y quebradizos. Uno de los problemas principales es la acumulación de hierro en el corazón y otros órganos, provocando insuficiencia cardiaca en algunos pacientes en los años de adolescencia o a principios de la década de los veinte.
-Beta talasemia leve o característica de talasemia - un gen beta tiene una deleción, provocando anemia. La talasemia leve se divide en:
1.-Talasemia mínima - la persona tiene pocos o ningún síntoma.
2.-Talasemia intermedia - la persona tiene una anemia de moderada a grave.
Las personas que tienen talasemia leve tienen un 50 por ciento de probabilidades de transmitirles el gen a sus hijos, quienes también tendrían talasemia leve. A muchas personas se les administran suplementos de hierro debido a la creencia errónea de que su anemia es del tipo ferropénico. Puesto que mucho hierro puede ser perjudicial, es importante consultar con un hematólogo acerca de cualquier tratamiento. La talasemia grave se hereda por un gen autosómico recesivo, lo que significa que las dos copias del gen son necesarias para producir la condición, una heredada de cada uno de los dos progenitores portadores que tienen talasemia leve.
Diagnóstico
La beta talasemia se encuentra con más frecuencia en personas de ascendencia mediterránea (griegos o italianos). Cada hijo de dos progenitores portadores tiene un 25 por ciento de probabilidades de padecer la enfermedad. El estado de portador puede determinarse por lo siguiente:
- Hemograma completo (su sigla en inglés es CBC) - medición del tamaño, el número y la madurez de diferentes glóbulos en un volumen específico de sangre.
- Electroforesis de la hemoglobina con cuantificación de A2 - procedimiento de laboratorio que diferencia los tipos de hemoglobina presentes.
Todos estos estudios pueden realizarse con una única muestra de sangre. El diagnóstico prenatal se determina a partir del muestreo de vellosidades coriales (su sigla en inglés es CVS) o de amniocentesis.
La transmisión es autosómica recesiva y se han identificado alrededor de 200 mutaciones (B0 o B+). Se recomienda el asesoramiento genético para permitir a las parejas en riesgo una elección informada entre las opciones disponibles, incluido el diagnóstico prenatal.
Tratamiento de la beta talasemia grave o anemia de Cooley
El tratamiento específico de la beta talasemia grave o anemia de Cooley será determinado por su médico basándose en lo siguiente:
- La edad de su hijo, su estado general de salud y sus antecedentes médicos.
- La gravedad de la enfermedad.
- La tolerancia de su hijo a determinados medicamentos, procedimientos o terapias.
- Sus expectativas para la evolución de la enfermedad
- Su opinión o preferencia.
El tratamiento para la beta talasemia puede incluir:
1.-Transfusiones de sangre regulares.
2.-Medicamentos (para disminuir la cantidad de hierro en el cuerpo, llamada terapia de quelación).
3.-extirpación quirúrgica del bazo (si fuera necesario)
4.-dosis diarias de ácido fólico
5.-Posible extirpación quirúrgica de la vesícula biliar.
6.-Ningún suplemento de hierro.
7.-Trasplante de médula ósea.
Véase también
Enlaces externos
| Control de autoridades |
|
|---|
-
 Datos: Q3616632
Datos: Q3616632
-
 Multimedia: Beta thalassemia / Q3616632
Multimedia: Beta thalassemia / Q3616632