
| Atrofia muscular espinal | ||
|---|---|---|
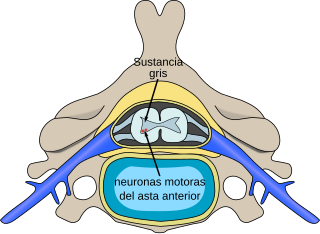 Localización, en la médula espinal, de neuronas afectadas por la atrofia muscular espinal
| ||
| Especialidad | neurología | |
| Síntomas | Debilidad muscular progresiva. | |
| Complicaciones | Escoliosis , contracturas articulares , neumonía | |
| Causas | Mutación en SMN1 | |
| Diagnóstico | Test genético | |
| Diagnóstico diferencial | Distrofia muscular congénita , distrofia muscular de Duchenne , síndrome de Prader-Willi | |
| Medicación | Nusinersen, Onasemnogene abeparvovec | |
La atrofia muscular espinal (AME) o enfermedad de la neurona motora inferior es un término aplicado a un variado número de trastornos que tienen en común una etiología genética y que se manifiestan como debilidad debida a lesiones de las neuronas motoras del asta anterior de la médula espinal, sobre todo las neuronas motoras inferiores del tallo encefálico y de la médula espinal.
Constituyen un grupo característico de enfermedades de la neurona motora de carácter autosómico recesivo que se inicia durante la infancia o la adolescencia. Por lo general se relacionan con alteraciones en el cromosoma 5, específicamente la deleción de un gen asociado a un 98% de los casos.
Se vincula la patología a un defecto en el gen SMN1, que es la copia telomérica codificante de la proteína de superveniencia de las neuronas motoras, y que da nombre al gen en inglés (survival of motor neuron 1, telomeric). El gen se sitúa en la molécula del genoma humano entre los pares de bases 70.924.941 a 70.953.015 del cromosoma 5. Tanto la copia telomérica como la centromérica (gen SMN2) de este gen están asociadas a la enfermedad, pero solo las mutaciones en la copia telomérica conducen a la enfermedad, mientras que la copia centromérica modula la enfermedad en caso de existir mutación en la telomérica. El SMN2 gen proporciona instrucciones para hacer varias versiones de la proteína SMN, pero solo una versión es funcional; las otras versiones son más pequeñas y se rompen fácilmente.
La enfermedad afecta a 1 cada 10 000 nacimientos. Se han identificado al menos 65 mutaciones en el SMN1 que causan la atrofia muscular espinal, y el 95 por ciento de los afectados tienen mutaciones que eliminan una sección del exón 7 en ambas copias del gen SMN1 en cada célula, en consecuencia no fabrican o apenas fabrican la proteína SMN. Entorno al 5 por ciento de los afectados tiene una copia del gen SMN1 con una deleción del exón 7, y la otra copia tiene una mutación diferente que interrumpe la producción o la función de la proteína SMN.
Histología
Al estudiar los músculos se aprecia una afectación de todo el fascículo muscular, lo que se denomina atrofia panfascicular. Se ven fibras hipertróficas que alcanzan de 2 a 4 veces el tamaño normal del músculo. Sin embargo, también se observa un gran número de fibras atróficas de pequeño diámetro.
Cuadro clínico
La atrofia muscular espinal infantil es la forma más grave y se acompaña de debilidad muscular, tono muscular disminuido, llanto débil y dificultad para tragar y para amamantar, lo que lleva a la acumulación de secreciones en los pulmones y la garganta y a una susceptibilidad a las infecciones respiratorias agudas. Físicamente se aprecia una notable tendencia a la atonía y alteraciones en la aparición de los reflejos infantiles normales.
Por lo general, cuanto más pronto aparecen los síntomas menor es la esperanza de vida del individuo. La aparición de la clínica es repentina y francamente notable, con rápido deterioro de las células neuronales motoras.
Tratamiento
Se han autorizado dos fármacos para el tratamiento de la enfermedad:
- Nusinersen. Aprobado por la FDA en diciembre de 2016.
- Onasemnogene abeparvovec. Aprobado por la FDA en mayo de 2019.
Tipos
- La AME tipo 1 aparece después del parto o en los primeros 6 meses de vida, y los pacientes pocas veces logran sobrevivir más de 3 años.
- La AME tipo 2 y 3 aparecen durante los primeros meses de vida o en la infancia, después de los 2 años. La supervivencia es más larga, en algunos casos hasta la vida adulta.
| Control de autoridades |
|
|---|
-
 Datos: Q580290
Datos: Q580290
-
 Multimedia: Spinal muscular atrophy / Q580290
Multimedia: Spinal muscular atrophy / Q580290