
| Astrocitoma pilocítico | ||
|---|---|---|
 Imagen de RM de un astrocitoma pilocítico en la región hipotalámica
| ||
| Especialidad | oncología | |
El astrocitoma pilocítico es el tumor glial más común en la edad pediátrica, con un pico de incidencia en los 10-12 años. También se diagnostica en adultos jóvenes menores de 20 años y sólo rara vez se da en los adultos mayores. De entre todos los astrocitomas, es el que tiene un pronóstico más favorable.
Este tumor se presenta generalmente como una lesión circunscrita y bien delimitada, de crecimiento lento y puede ser sólido o quístico. Suelen apreciarse con facilidad en las imágenes de TC y RM. Los quistes pueden ser monoloculares o multiloculares, con el nódulo tumoral encapsulado en el interior. El astrocitoma pilocítico generalmente aparece a nivel del cerebelo, tronco cerebral, región hipotalámica, nervios y quiasma óptico, y en general en cualquier área donde haya presencia de astrocitos, incluyendo los hemisferios cerebrales y la médula espinal. La localización más frecuente es el cerebelo.
Estos tumores suelen ser no infiltrantes, en el sentido de que tienden a permanecer en el área donde se iniciaron y no se diseminan al tejido circundante, por lo que se clasifican en el grado I de malignidad de la OMS. Su progresión a grados superiores es extremadamente rara. En muchos casos, la cirugía puede ser curativa si se consigue una resección completa del tumor.

Epidemiología
El astrocitoma pilocítico es el glioma más frecuente en niños, con una incidencia en ambos sexos de entre 0,5 y 0,6 casos por 100 000/año. Después de la segunda década de edad la incidencia disminuye, con sólo unos pocos casos presentes en mayores de 50 años. Este tumor representa el 10 % de los astrocitomas cerebrales y el 85 % de los cerebelosos.
Etiología y patogénesis
A día de hoy no se conocen con exactitud los factores que desencadenan la aparición del astrocitoma pilocítico. A menudo se le ha asociado con la neurofibromatosis tipo 1 (NF1), una enfermedad autosómica dominante caracterizada, entre otras cosas, por el desarrollo de tumores de pronóstico más o menos favorable. Los gliomas de las vías ópticas, de los cuales el 60 % son astrocitomas pilocíticos, son tumores comunes en pacientes con esta enfermedad.
Anatomía patológica
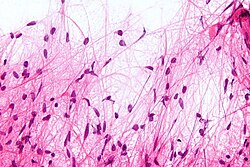
Macroscópicamente, el astrocitoma pilocítico suele presentarse como una masa quística bien definida, con un nódulo mural en la pared del quiste. Si es sólido, puede estar bien circunscrito o, con menos frecuencia, ser infiltrativo.
Microscópicamente, el tumor muestra un patrón bifásico, con células bipolares compactas de forma pilosa (de ahí su denominación "pilocítico") con largos procesos positivos para GFAP y zonas hipocelulares con microquistes. En las zonas sólidas, la neoplasia se compone de astrocitos neoplásicos con un citoplasma fibrilar intensamente eosinofílico y núcleos alargados y redondeados sin atipias. En las zonas microquísticas las prolongaciones citoplasmáticas tienen siempre la apariencia de fibrillas pero son menores en número y se irradian en todas direcciones. Son muy frecuentes en el astrocitoma pilocítico las fibras de Rosenthal y los cuerpos granulares eosinofílicos. También puede haber proliferación vascular de aspecto glomeruloide no indicativa de malignidad.
Signos y síntomas
Los síntomas más comunes son debidos a la compresión del parénquima cerebral como resultado del efecto masa (debido a la hiperactividad de la proliferación celular) y al aumento de la presión intracraneal. Los síntomas pueden variar dependiendo de la ubicación y el tamaño de la neoplasia, aunque los más frecuentes son náuseas, vómitos, dolor de cabeza, ataxia y alteraciones visuales.
Diagnóstico
El diagnóstico de este tumor se lleva a cabo normalmente usando la tomografía computarizada (TC) o la resonancia magnética nuclear (RM) del cráneo, con medio de contraste. De manera complementaria, puede añadirse una tomografía por emisión de positrones (PET).
La RM ofrece una mayor sensibilidad en comparación con la TC, si bien no siempre es fácilmente accesible y presenta contraindicaciones, por ejemplo, no se puede utilizar en pacientes con marcapasos, prótesis incompatibles con el campo magnético, clips metálicos, etc. En las imágenes de TC y RM el astrocitoma pilocítico aparece como un tumor con márgenes bien definidos, redondeado, sólido o asociado a macro o microquistes. El 10 % presenta calcificación. La captación de contraste es prácticamente homogénea y muy intensa. Este hallazgo es indicativo de una fuerte vascularización del tumor, aunque en este caso no se considera indicativa de malignidad. En fosa posterior, el diagnóstico diferencial se hace con el meduloblastoma, el ependimoma y el hemangioblastoma, y en la región quiasmática con el craneofaringioma.
Tratamiento

El tratamiento de elección suele ser la cirugía. A menudo es posible una resección completa del tumor, aunque esto depende de su localización y de su facilidad de acceso. La remoción radical del tumor, si es posible, es curativa y generalmente permite la supervivencia funcional durante muchos años.
En pacientes con una resección incompleta, el curso clínico es a menudo benigno y suele conseguirse una estabilización postoperatoria de la enfermedad, a pesar de la presencia de restos tumorales en los márgenes quirúrgicos. Por esta razón, el uso de radioterapia postoperatoria en estos pacientes es controvertido. En los raros casos de recidivas a distancia, la primera opción de tratamiento es una segunda intervención quirúrgica. En los casos de segundas recidivas, se recomienda al paciente tratamiento radioterápico o quimioterápico.
Seguimiento
Después de la cirugía, el paciente debe ser controlado con RM encefálica cada seis meses durante los dos primeros años, y después anualmente durante otros 5-6 años. Finalmente, la frecuencia de los controles de imagen pasa a ser cada dos o tres años.
Pronóstico
El astrocitoma pilocítico es el astrocitoma con mejor pronóstico. Si se realiza una resección total, la supervivencia a los 10 años es aproximadamente del 90 % y después de una resección parcial, la supervivencia a los 10 años se sitúa aproximadamente en el 45 %. La enfermedad tiene un pronóstico generalmente benigno y muy rara vez se detecta una transformación maligna con mal pronóstico. En la literatura se han descrito casos excepcionalmente raros de anaplasia, hasta el punto de sugerir para ellos la denominación de astrocitoma pilocítico anaplásico (APA), con un comportamiento apenas distinguible al del glioblastoma.
Bibliografía
- P.C. Burger et al., Pilocitic Astrocytoma. In Kleihues P, Cavenee WK, eds., Pathology and genetics of tumours of the nervous system, World Health Organization classification of tumours. Lyon, France: IARC Press, 2000, ISBN 9283224094
- Lisa M. DeAngelis (2001). Brain Tumors, N Engl J Med, Vol. 344(2):114-123, January 11, 2001.
- Escalona-Zapata, Julio; Bello, M. J. (1996). Tumores del sistema nervioso central. Editorial Complutense. ISBN 8489365598.
- Reyes Oliveros, Francisco (2007). Gliomas del encéfalo. Univ. Santiago de Compostela. ISBN 8497508130.
- Aguirre Cruz, María Lucinda; Sotelo Morales, Julio (2008). Tumores cerebrales. Volumen 1. Ed. Médica Panamericana. ISBN 9687988843.
Enlaces externos
-
 Wikimedia Commons alberga una categoría multimedia sobre Astrocitoma pilocítico.
Wikimedia Commons alberga una categoría multimedia sobre Astrocitoma pilocítico. - Astrocitomas infantiles - Instituto Nacional del Cáncer de Estados Unidos
-
 Datos: Q2095252
Datos: Q2095252
-
Multimedia: Pilocytic astrocytoma / Q2095252