

El aprendizaje automático en bioinformática consiste en la aplicación de algoritmos de aprendizaje automático, en entornos de bioinformática, como, por ejemplo, la genómica, la proteómica, los microarrays, la biología de sistemas, la biología evolutiva y la minería de textos. Esto permite automatizar la búsqueda de patrones complejos en series de datos, facilitando la comprensión de procesos biológicos tan complejos como la estructura de las proteínas, lo que diferencia a esta disciplina de los enfoques tradicionales de bioinformática, que requieren supervisión y que dificultan la aparición de patrones inesperados u ocultos.
Se trata de un campo emergente debido al gran crecimiento de la cantidad de conjuntos de datos biológicos disponibles, necesarios para entrenar algoritmos con un nivel de detalle alto.
Funcionamiento
Los algoritmos de aprendizaje automático en bioinformática pueden utilizarse para la predicción, la clasificación y la selección de características. Los métodos para lograr esta tarea son variados y abarcan muchas disciplinas; los más conocidos son el aprendizaje automático y la estadística. Las tareas de clasificación y predicción tienen como objetivo construir modelos que describan y distingan clases o conceptos para su futura predicción. Las diferencias entre ellas son las siguientes:
- La clasificación produce una variable discreta, mientras que la predicción produce una característica de valor numérico.
- El tipo de algoritmo o proceso utilizado para construir los modelos de predicción a partir de los datos utilizando analogías, reglas, redes neuronales, probabilidades y/o estadísticas.
Debido al crecimiento exponencial de las tecnologías de la información y de los modelos aplicables, incluyendo la inteligencia artificial y la minería de datos, además del acceso a conjuntos de datos cada vez más amplios, se han creado nuevas y mejores técnicas de análisis de la información, basadas en su capacidad de aprendizaje. Dichos modelos permiten ir más allá de la descripción y proporcionan conocimientos en forma de modelos comprobables. Algunos de los enfoques más usados son:
Clasificación
En este tipo de tareas de aprendizaje automático, la salida es una variable discreta. Un ejemplo de este tipo de tarea en bioinformática es el etiquetado de nuevos datos genómicos (como genomas de bacterias no cultivables) basado en un modelo de datos ya etiquetados.
Modelos ocultos de Markov
Los modelos ocultos de Markov son un tipo de modelos estadísticos para datos secuenciales. Un Modelo de Markov está compuesto por dos objetos matemáticos: un proceso observado dependiente del estado , y un proceso de estado no observado u oculto. Así, el proceso en estudio no se observa directamente, sino que se realizan observaciones sobre un proceso dependiente del proceso de interés, y cuyos cambios sirven como una medida ruidosa de los estados del sistema de interés. Así, pueden utilizarse para perfilar y convertir un alineamiento de secuencias múltiples en un sistema de puntuación específico para cada posición, adecuado para buscar secuencias homólogas en las bases de datos de forma remota.
Redes neuronales convolucionales
Las redes neuronales convolucionales son una clase de redes neuronales profundas cuya arquitectura se en una serie de perceptores con varias capas, que posteriormente se conectan entre sí. Estas redes fueron inspiradas por procesos biológicos en los que el patrón de conectividad entre neuronas se asemeja a la organización de la corteza visual animal, y utilizan relativamente poco preprocesamiento en comparación con otros algoritmos. Así, la red aprende a optimizar sus filtros a través del aprendizaje automatizado, mientras que en los algoritmos tradicionales requieren que estos filtros sean diseñados a mano.
Bosque aleatorio
Los bosques aleatorios clasifican construyendo un conjunto de árboles de decisión, y emitiendo la predicción media de los árboles individuales. Desde el punto de vista computacional, los bosques aleatorios son atractivos porque manejan de forma natural tanto la regresión como la clasificación, son relativamente rápidos de entrenar y predecir, dependen sólo de uno o dos parámetros de ajuste, tienen una estimación incorporada del error de generalización, pueden utilizarse directamente para problemas de alta dimensión y pueden implementarse fácilmente en paralelo. Desde el punto de vista estadístico, los bosques aleatorios son atractivos para las características adicionales, como las medidas de importancia de las variables, la ponderación diferencial de las clases, la imputación de valores perdidos, la visualización, la detección de valores atípicos y el aprendizaje no supervisado.
Agrupamiento
La agrupación (clustering) de un conjunto de datos en subconjuntos, de modo que los datos de cada subconjunto estén lo más cerca posible entre sí y lo más distantes posible de los datos de cualquier otro subconjunto, es una técnica común para el análisis estadístico de datos. El clustering es fundamental para muchas investigaciones bioinformáticas, y sirve como un poderoso método computacional para analizar datos no estructurados y de alta dimensión en forma de secuencias, expresiones, textos, imágenes, etc. El clustering también se utiliza para obtener información sobre los procesos biológicos a nivel genómico, por ejemplo, las funciones de los genes, los procesos celulares, los subtipos de células, la regulación génica y los procesos metabólicos. Existen dos tipos de algoritmos de agrupamiento, a grandes rasgos:
- Los algoritmos jerárquicos encuentran grupos usando grupos previamente establecidos, de forma aglomerativa (bottom-up, poniendo cada elemento en un grupo propio, y fusionándolos sucesivamente en otros más grandes) o divisiva (top-down, dividiendo el conjunto en subgrupos). Un ejemplo es BIRC, que es particularmente bueno en bioinformática por su complejidad de tiempo casi lineal dado conjuntos de datos generalmente grandes.
- Los algoritmos particionales determinan todos los clusters a la vez, pues se basan en especificar un número inicial de grupos, y reasignar iterativamente los objetos entre los grupos hasta la convergencia; por ejemplo, k-means o k-medoids.
Aplicaciones
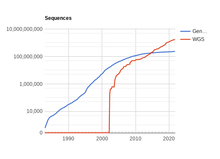
Genómica
Aunque los datos de genómica han sido históricamente escasos debido a la dificultad técnica de secuenciar un fragmento de ADN, en los últimos años el número de secuencias indexadas en bases de datos está creciendo exponencialmente. Sin embargo, la interpretación de estos datos está ocurriendo a un ritmo mucho más lento, por lo que existe una necesidad creciente de desarrollar sistemas que puedan determinar automáticamente qué partes de una secuencia de ADN dada codifican proteínas y cuáles no: se trata de la predicción de gen.
Esta predicción se puede realizar mediante búsqueda extrínseca e intrínseca:
- En la extrínseca, una secuencia de ADN se pasa por una base de datos de secuencias anotadas, buscando homólogos a genes ya existentes
- En la intrínseca, se buscan regiones codificantes en la cadena "por sí misma", sin ayuda de agentes externos.
El aprendizaje automático también puede usarse para la alineación de secuencias, que junta regiones de semejanza para detectar una historia evolutiva compartida, así como regiones recombinantes.
Metagenómica
La metagenómica es un campo nuevo de investigación que se ha desarrollado en la última década, con el objeto de entender la diversidad y le riqueza microbiana de diferentes nichos ecológicos conformados por microorganismos cultivables y no cultivables, teniendo así aplicaciones en diferentes campos de la investigación médica, sistemas agropecuarios, estudios medioambientales y de sostenibilidad, y en la industria.
La metagenómica es el estudio de comunidades microbianas de un entorno, por medio de muestras de ADN. Esta conlleva a la extracción, clonación, secuenciación y análisis del genoma de una comunidad microbiana, lo cual permite el estudio de una gran variedad de genes y sus productos; esto nos puede proporcionar información taxonómica e incluso metabólica o funcional de las comunidades estudiadas.
Actualmente, en el estudio metagenómico bioinformático predominan las limitaciones y desafíos en la implementación de herramientas de aprendizaje automático debido a la cantidad de datos en las muestras ambientales, ya que al tomar una muestra de un ambiente, esta contienen genes de todo organismo perteneciente a este entorno; esto lleva a la alta dimensionalidad de los conjuntos de datos del microbioma.
Proteómica
Las proteínas son cadenas de aminoácidos cuya funcionalidad depende en gran medida de su estructura tridimensional. Esta estructura se va "plegando" progresivamente, pasando desde la estructura primaria (la cadena lineal de aminoácidos) a la estructura secundaria que predice la terciaria, y si se asocia con otras cadenas peptídicas, la cuaternaria. Así, la predicción de la estructura secundaria directamente desde la primaria es un campo con grandes aplicaciones en el área de la farmacodinámica, aunque se trata de un proceso increíblemente costoso y que requiere mucho tiempo, y que, hasta la llegada del aprendizaje automático, debía realizarse manualmente.
Hoy en día, mediante el uso del aprendizaje automático se puede alcanzar una precisión del 82-84%, usando redes neuronales artificiales para clasificar regiones del ADN en función de si codifican para una hélice alfa, lámina beta o espiral). El aprendizaje automático también se puede usar para la predicción de cadenas laterales, el modelado de bucles de proteínas, y la generación de mapas de contacto.
Microarrays
Los microarrays son un tipo de chip de laboratorio utilizado para recopilar datos de forma automática sobre grandes cantidades de material biológico. Esta tecnología permite controlar la expresión diferencial de genes, ayudando a diagnosticar enfermedades como el cáncer. Sin embargo, debido a la enorme cantidad de datos recopilados, se hace necesario discernir cuales son de interés y cuales no para el diagnóstico, para lo que se pueden usar métodos de aprendizaje automático tales como el aprendizaje profundo o los árboles de decisión.
Biología de sistemas
La biología de sistemas se centra en el estudio de las interacciones complejas de componentes biológicos simples en un sistema. Por ejemplo, se pueden modelar redes génicas y estructuras reguladoras a partir de la expresión de los genes individuales usando modelos gráficos probabilistas, identificar sitios de unión de factores de transcripción utilizando la optimización de cadena de Markov o identificar la sensibilidad de los genes NCR (encargados de desatar la citotoxicidad) en levadura.
Minería de textos

El aumento de las publicaciones científicas dificulta la búsqueda y compilación de toda la información relevante disponible sobre un tema determinado. Esto, a su vez, dificulta la recopilación de datos biológicos necesaria para todas las aplicaciones anteriormente mencionadas. El procesamiento del lenguaje natural permite extraer la información útil dentro de los informes generados por humanos en una base de datos dada. Además, como las anotaciones de proteínas en las bases de datos de proteínas a menudo no reflejan el conjunto de conocimiento conocido, se puede extraer información adicional de la literatura biomédica, permitiendo funciones como la anotación automática, la determinación de la localización subcelular de una proteína, el análisis de interacción de proteínas a gran escala o la detección y visualización de regiones que comparten un alto grado de semejanza.
Bases de datos
Una parte importante de la bioinformática es la gestión de grandes conjuntos de datos, conocidos como bases de datos de referencia. Existen bases de datos para cada tipo de datos biológicos, por ejemplo para los grupos de genes biosintéticos y los metagenomas.
- Centro Nacional de Información Biotecnológica: El NCBI ofrece un amplio conjunto de recursos en línea de información y datos biológicos, como la base de datos de secuencias de ácidos nucleicos GenBank y la base de datos de citas y resúmenes de revistas de ciencias de la vida PubMed. Muchas de las aplicaciones web se complementan con implementaciones personalizadas del programa BLAST, optimizadas para la búsqueda de conjuntos de datos especializados. Los recursos incluyen la gestión de datos de PubMed, los elementos funcionales de RefSeq, la descarga de datos del genoma, la API de servicios de variación, Magic-BLAST, QuickBLASTp y los grupos de proteínas idénticas. Se puede acceder a todos estos recursos a través del NCBI.
- antiSMASH: Permite la rápida identificación, anotación y análisis de grupos de genes de biosíntesis de metabolitos secundarios en genomas bacterianos y fúngicos. Se integra y cruza con un gran número de herramientas de análisis in silico de metabolitos secundarios.
- gutSMASH: Evalúa sistemáticamente el potencial metabólico bacteriano mediante la predicción de clústeres de genes metabólicos (MGC), tanto conocidos como novedosos, del microbioma intestinal.
- MIBiG: Son las siglas de Minimal Identifier for Biosintetic Gene cluster, y proporciona un estándar para las anotaciones y metadatos sobre los clústeres de genes biosintéticos y sus productos moleculares. Se trata de un proyecto del Consorcio de Estándares Genómicos que se basa en el marco de información mínima sobre cualquier secuencia (MIxS).
- SILVA: es un proyecto interdisciplinario entre biólogos e informáticos que reúne una base de datos completa de secuencias de genes de ARN ribosómico (ARNr), tanto de las subunidades pequeñas (16S,18S, SSU) como de las grandes (23S, 28S, LSU), que pertenecen a los dominios de las bacterias, las arqueas y las eucariotas. Estos datos están disponibles gratuitamente para uso académico y comercial.
- Greengenes: es una base de datos de genes de ARN ribosomal 16S de longitud completa que proporciona cribado de quimeras, alineación estándar y una taxonomía curada basada en la inferencia de árboles de novo.
- Open Tree of life Taxonomy: Tiene como objetivo construir un Árbol de la Vida completo, dinámico y disponible digitalmente, sintetizando los árboles filogenéticos publicados junto con los datos taxonómicos. Aunque menos utilizada, tiene un mayor número de secuencias clasificadas taxonómicamente hasta el nivel de género en comparación con SILVA y Greengenes.
- Ribosomal Database Project: Es una base de datos que proporciona secuencias de ARN ribosómico (ARNr) de subunidades pequeñas de dominio bacteriano y arqueológico (16S); y secuencias de ARNr fúngico de subunidades grandes (28S).
Enlaces externos
- Esta obra contiene una traducción derivada de «Machine learning in bioinformatics» de Wikipedia en inglés, concretamente de esta versión, publicada por sus editores bajo la Licencia de documentación libre de GNU y la Licencia Creative Commons Atribución-CompartirIgual 3.0 Unported.
| Control de autoridades |
|
|---|
-
 Datos: Q30314784
Datos: Q30314784