
Las tecnologías de transcriptómica son las técnicas utilizadas para el estudio del transcriptoma de un organismo, es decir, el conjunto de todos sus transcritos de ARN. La información contenida en un organismo se guarda en el ADN de su genoma y se expresa mediante la transcripción. Entonces, el ARNm sirve como una molécula intermediaria y transitoria en la red de información, mientras que los ARNs no codificantes cumplen con diferentes funciones adicionales. Un transcriptoma refleja una foto fija en el tiempo de los transcritos totales presentes en una célula. Las tecnologías de transcriptómica permiten saber qué procesos celulares se encuentran activos y cuáles inactivos. Uno de los principales retos de la biología molecular es comprender cómo un único genoma origina variedad de células. Otro es cómo se regula la expresión génica.
Los primeros intentos en estudiar transcriptomas completos comenzó al comienzo de la década de 1990. Desde finales de esta década, los subsiguientes avances tecnológicos han transformado repetidamente el campo y hecho de la transcriptómica una disciplina generalizada en las ciencias biológicas. Existen dos técnicas actuales clave en este campo: microarrays, los cuales cuantifican un conjunto de secuencias predeterminadas, y ARN-Seq, el cual utiliza secuenciación de alto rendimiento para registrar todos los transcritos. Conforme la tecnología ha mejorado, el volumen de datos producidos por cada experimento de transcriptómica se ha incrementado. Como resultado, los métodos de análisis de datos se han adaptado continuamente para analizar, de manera más precisa y eficiente, grandes volúmenes de datos. Las bases de datos transcriptómicos también se han ampliado y se han vuelto más útiles a medida que los investigadores continúan coleccionando y compartiendo transcriptomas. Sin el conocimiento de experimentos previos, sería imposible interpretar la información contenida en un transcriptoma.
La medición de la expresión de los genes de un organismo en tejidos, condiciones o momentos diferentes, aporta información sobre cómo los genes se regulan y revela detalles de la biología de un organismo. También se puede utilizar para inferir las funciones de genes previamente no anotados. Los análisis del transcriptoma han permitido el estudio de cómo la expresión génica cambia en diferentes organismos y ha sido crucial en la comprensión de las enfermedades humanas. Un análisis completo de la expresión génica hace posible la detección de tendencias amplias y coordinadas que no pueden discernirse de otra forma mediante ensayos más específicos.
Historia
La transcriptómica se ha caracterizado por el desarrollo de nuevas técnicas, las cuales han redefinido cada década lo que es posible y han convertido en obsoletas tecnologías previas. El primer intento en capturar el transcriptoma humano de manera parcial fue publicado en 1991 e informó de 609 secuencias de ARNm del cerebro humano. En 2008 se publicaron dos transcriptomas humanos, compuestos de millones de secuencias derivadas de transcritos y cubriendo 16 000 genes. En 2015, se habían publicado transcriptomas de cientos de individuos. Actualmente, se generan continuamente transcriptomas de diferentes enfermedades, tejidos o incluso de células únicas. El rápido desarrollo de nuevas tecnologías con sensibilidad mejorada y más baratas ha posibilitado esta explosión en transcriptómica.
Antes de la transcriptómica
Varias décadas antes de que estuviera disponible cualquier estrategia de transcriptómica, ya se realizaban estudios de transcrítos individuales. A finales de la decada de 1970, se coleccionaron transcritos de ARNm de la mosca de seda, convirtiéndolos a ADN complementario (ADNc) para almacenamiento mediante transcriptasa inversa. En los años 1980, la secuenciación de bajo rendimiento utilizando el método de Sanger se utilizó para secuenciar transcritos al azar, produciendo marcadores de secuencias expresadas (ESTs). El método de secuenciación de Sanger predominaba hasta la llegada de los métodos de alto rendimiento, tales como la secuenciación por síntesis (Solexa/Illumina). Las secuencias ESTs se convirtieron en las predominantes durante los años 1990 como un método eficiente para determinar el contenido de genes de un organismo sin secuenciar el genoma completo. Multitud de métodos permitieron cuantificar transcritos individuales, tales como northern blot, matrices de membrana de nylon y PCR cuantitativa tras transcriptasa inversa (RT-qPCR).Sin embargo, estos métodos son laboriosos y solo pueden capturar una pequeña subsección del transcriptoma. Consecuentemente, la manera en que un transcriptoma en su conjunto se expresa y regula permaneció desconocida hasta el desarrollo de las técnicas de alto rendimiento.
Primeros intentos
El término transcriptoma se utilizó por primera vez en la década de los años 1990. En 1995, se desarrolló uno de los primeros métodos de secuenciación basados en transcriptómicas , el análisis seriado de expresión génica (SAGE), la cual funcionaba mediante secuenciación de Sanger de fragmentos transcritos concatenados al azar. Los transcritos se cuantificaron comparando los fragmentos con genes conocidos. También se utilizó en un breve espacio de tiempo una variante de SAGE, que utiliza técnicas de secuenciación de alto rendimiento, denominada análisis de expresión génica digital. Sin embargo, estos métodos fueron rápidamente reemplazados por la secuenciación de alto rendimiento de transcritos completos, lo cual proveía de información adicional sobre la estructura de los transcritos, por ejemplo, variantes de empalme alternativo.
Desarrollo de técnicas contemporáneas.
| ARN-Seq | Microarray | |
|---|---|---|
| Rendimiento | 1-7 días por experimento | 1-2 días por experimento |
| Cantidad de ARN de entrada | Bajo ~ 1 ng de ARN total | Alto ~ 1 μg de ARNm |
| Intensidad de trabajo | Alto (preparación de muestras y análisis de datos) | Bajo |
| Conocimiento previo | No se requiere ninguno, aunque conocer una secuencia de genoma/transcriptoma de referencia es útil | Se requiere un genoma/transcriptoma de referencia para el diseño de sondas |
| Precisión en la cuantificación | ~90 % (limitado por la cobertura de la secuencia) | >90 % (limitado por la precisión de detección de fluorescencia) |
| Resolución de secuencia | ARN-Seq puede detectar SNPs y variantes de empalme (limitado por la precisión de secuenciación de ~99 %) | Las matrices especializadas pueden detectar variantes de empalme de ARNm (limitado por el diseño de la sonda y la hibridación cruzada) |
| Sensibilidad | 1 transcrito/millón (aproximado, limitado por la cobertura de la secuencia) | 1 transcrito/mil (aproximado, limitado por detección de fluorescencia) |
| Rango dinámico | 100 000:1 (limitado por la cobertura de secuencia) | 1000:1 (limitado por la saturación de fluorescencia) |
| Reproducibilidad técnica | >99 % | >99 % |
Las técnicas predominantes actualmente, los microarrays y el ARN-Seq, se desarrollaron en la mitad de la década de 1990 y de 2000. En 1995 se publicaron por primera vez microarrays capaces de medir la abundancia de un conjunto definido de transcritos mediante su hibridación en una matriz de sondas complementarias. La tecnología de microarray permitió el ensayo de miles de transcritos simultáneamente y a un coste considerablemente reducido por gen y con ahorro de trabajo manual. Tanto las matrices de oligonucleótidos individuales como las de alta densidad de Affymetrix fueron los métodos preferidos para la elaboración de perfiles transcripcionales hasta finales de la década de 2000. Durante este periodo, se produjeron una serie de microarrays para abarcar genes conocidos de organismos modelo o de importancia económica. Los avances en el diseño y fabricación de matrices mejoraron la especificidad de las sondas y permitieron analizar más genes en un única matriz. Los avances en la detección por fluorescencia aumentaron la sensibilidad y la precisión de las mediciones de transcritos de baja abundancia.
El ARN-Seq se realiza mediante la transcripción inversa in vitro del ARN y la secuenciación del ADNc resultante. La abundancia de los transcritos se obtiene a partir del número de recuentes de cada transcrito. Por lo tanto, la técnica está altamente influenciada por el desarrollo de las tecnologías de secuenciación de alto rendimiento. La secuenciación masiva en paralelo (MPSS) fue un ejemplo temprano basado en la generación de secuencias de 16-20 pares de base mediante series complejas de hibridaciones, y se utilizó en 2004 para validar la expresión de 10 000 genes en Arabidopsis thaliana. El primer proyecto de ARN-Seq se publicó en 2006 con 1000 transcritos secuenciados utilizando tecnología 454. Esto fue suficiente cobertura para cuantificar la abundancia relativa de los transcritos. El ARN-Seq comenzó a ser a popular después de 2008 cuando nuevas tecnologías Solexa/Illumina permitieron registrar mil millones de secuencias de transcritos. Este rendimiento permite ahora la cuantificación y comparación de transcriptomas humanos.
Recopilación de datos
La generación de datos sobre los transcritos de ARN es posible mediante dos aproximaciones fundamentales: secuenciación de transcritos individuales (ESTs, o ARN-Seq) o hibridación de una matriz ordenada de sondas de nucleótidos (microarrays).
Aislamiento de ARN
Todos los métodos de transcriptómica requieren primero el aislamiento del ARN del organismo experimental antes de poder registrar los transcritos. Aunque los sistemas biológicos son increíblemente diversos, las técnicas de extracción de ARN son muy similares entre sí e involucran la disrupción mecánica de células y tejidos, disrupción de ARNasa con sales caotrópicas, disrupción de macromoléculas u complejos de nucleótidos, separación del ARN de biomoléculas indeseadas incluyendo ADN, y concentración del ARN mediante precipitación de una solución o dilución de una matriz sólida. El ARN aislado puede ser tratado adicionalmente con DNasa para digerir cualquier traza de ADN. Es necesario el enriquecimiento del ARN mensajero, ya que los extractos de ARN total, típicamente, se componen de ARN ribosómico en un 98%. El enriquecimiento de transcritos se puede llevar a cabo mediante métodos de afinidad por la poli-A o por depleción de ARN ribosómico mediante sondas específicas de secuencia. El ARN degradado puede afectar a los resultados posteriores; por ejemplo, el enriquecimiento de ARNm a partir de muestras degradas dará como resultado la depleción de los extremos 5' del ARNm y una señal irregular a lo largo de la longitud de cada transcrito. Es típica la congelación rápida de tejidos previa al aislamiento del ARN. Así se reduce la exposición de las enzimas RNasa una vez que el aislamiento está completo.
Marcadores de secuencia expresada
Un marcador de secuencia expresada (EST) es una secuencia corta de nucleótidos generada a partir de un solo transcrito de ARN. El ARN se copia primero en forma de ADN complementario (ADNc) mediante una enzima transcriptasa inversa, antes de la secuenciación del ADNc resultante. Ya que los ESTs se pueden coleccionar sin el conocimiento previo del organismo de procedencia, se pueden crear a partir de mezclas de diferentes organismos de muestras ambientales. Aunque actualmente se utilizan métodos de mayor rendimiento, las bibliotecas de ESTs solían aportar información de secuencias para diseños tempranos de microarrays; por ejemplo, se diseñó un microarray de cebada a partir de 350 000 ESTs secuenciados.
Análisis en serie y cap de la expresión génica (SAGE/CAGE)

El análisis en serie de la expresión génica (SAGE) se desarrolló a partir de la metodología de ESTs para incrementar el rendimiento de los marcadores generados y permitir la cuantificación de la abundancia de transcritos. El ADNc se genera a partir del ARN, pero luego se digiere en fragmentos "marcadores" de 11 pares de bases, mediante enzimas de restricción que cortan el ADN en secuencias específicas. Entonces, las secuencias se dividen de nuevo en sus fragmentos originales de 11 pares de bases mediante un software de computación, en un proceso llamado deconvolución. Si existe un genoma de referencia de alta calidad, estos marcadores se pueden emparejar con su gen correspondiente en el genoma. Si no existe un genoma de referencia, los marcadores se pueden utilizar directamente como marcadores diagnósticos si se encuentran expresados diferencialmente en un estado de enfermedad.
El análisis en cap de la expresión génica (CAGE) es un método derivado del SAGE que secuencia marcadores solo desde el extremo 5' de un transcrito de ARNm. Por lo tanto, el sitio de inicio de la transcripción de los genes se puede identificar cuando los marcadores se alinean a un genoma de referencia. La identificación de sitios de inicio en los genes es útil para el análisis de promotores y para la clonación de ADNc en su longitud completa.
SAGE y CAGE son métodos que generan información de más genes de los que era posible mediante la secuenciación de ESTs únicos, pero la preparación de las muestras y el análisis de datos son típicamente más intensivos.
Microarrays
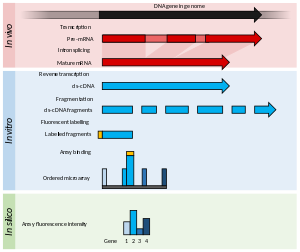
Principios y avances
Los microarrays consisten normalmente de una gradilla de oligómeros cortos de nucleótidos, llamados sondas y típicamente organizados en un portaobjetos de vidrio. La abundancia de transcritos se determina por hibridación de los transcritos marcados por fluorescencia a estas sondas. La intensidad de la fluorescencia en cada sonda del array indica la abundancia del transcrito para la secuencia de esa sonda. Se puede diseñar grupos de sondas para medir el mismo transcrito (por ejemplo, hibridando un transcrito en específico en diferentes posiciones) y suelen denominarse "conjuntos de sondas".
Los microarrays necesitan cierto conocimiento sobre la genómica del organismo de interés, por ejemplo, en forma de una secuencia del genoma anotada, o una genoteca de ESTs que se pueda utilizar para generar las sondas para la matriz.
Métodos
Los microarrays utilizados en transcriptómica normalmente se clasifican en dos amplias categorías: matrices punteadas de baja densidad o matrices de sondas pequeñas y alta densidad. La abundancia de transcritos se infiere por la intensidad de la fluorescencia, producto de transcritos marcados con fluoróforos, que se unen a la matriz.
Las matrices punteadas de baja densidad muestran normalmente gotas de picolitro de un rango de ADNc purificado en la superficie de un portaobjetos de vidrio. Estas sondas son más largas que las utilizadas en matrices de alta densidad y no pueden identificar eventos de empalme alternativo. Las matrices punteadas usan dos fluoróforos diferentes para marcar las muestras test y control, y el ratio de fluorescencia se utiliza para calcular de manera relativa la abundancia. Por otra parte, los de alta densidad utilizan un único marcador fluorescente, y cada muestra se hibrida y detecta individualmente. Estos arrays se popularizaron por el array Affymetrix GeneChip, en el que cada transcrito se cuantifica por diferentes sondas cortas de 25 oligómeros que analizan conjuntamente un gen.
Las matrices NimbleGen fueron un modelo de matriz de alta densidad producidas por un método de fotoquímica sin máscara, el cual permitió la manufactura flexible de matrices tanto en cantidades pequeñas como grandes. Estas tenían 100 000s de sondas de 45 a 85 oligómeros y se hibridaban con una muestra marcada de un color para el análisis de la expresión. Algunos diseños incorporaban hasta 12 matrices independientes por portaobjetos.
ARN-Seq

Principios y avances
El ARN-Seq consiste en la combinación de metodologías de secuenciación de alto rendimiento con métodos computacionales para capturar y cuantificar transcritos presentes en un extracto de ARN. La secuencia de nucleótidos generada es normalmente de una longitud de 100 pares de bases, aunque puede variar desde 30 a más de 10 000 pares de bases dependiendo del método de secuenciación utilizado. El ARN-Seq aprovecha el muestreo profundo del transcriptoma con muchos fragmentos pequeños de este para permitir la reconstrucción computacional del transcrito de ARN original alineando las lecturas a un genoma de referencia o entre sí (ensamblaje de novo). En un experimento de ARN-Seq se pueden cuantificar tanto los ARNs de baja como de alta abundancia (rango dinámico de 5 ordenes de magnitud) - una ventaja clave frente a los transcriptomas mediante microarray. Además, la cantidad de ARN de entrada es mucho menor para ARN-Seq (cantidades de nanogramos) en comparación con los microarrays (cantidades de microgramos), lo cual permite un examen más fino de las estructuras celulares hasta el nivel unicelular cuando se combina con la amplificación lineal de ADNc. En teoría, no existe un límite máximo para la cuantificación por ARN-Seq, y el ruido de fondo es muy bajo para lecturas de 100 pares de bases en regiones no repetitivas.
El ARN-Seq se puede utilizar para identificar genes en un genoma o para identificar qué genes están activos en un momento concreto en el tiempo. El conteo de lecturas se puede utilizar para modelizar de manera precisa los niveles relativos de expresión génica. La metodología del ARN-Seq ha mejorado continuamente, principalmente por el desarrollo de tecnologías de secuenciación de ADN para incrementar el rendimiento, precisión y longitud de lectura. Desde las primeras descripciones en 2006 y 2008, el ARN-Seq se ha adoptado rápidamente y superó a los microarrays como la técnica dominante en transcriptómica en 2015.
El objetivo de generar datos transcriptómicos a nivel de células individuales ha impulsado avances en los métodos de preparación de genotecas para ARN-Seq, dando lugar a avances espectaculares en sensibilidad. En la actualidad, los transcriptomas de célula única están bien descritos y incluso se han extendido a ARN-Seq in situ, en el que los transcriptomas de células individuales se analizan directamente en tejidos fijados.
Métodos
El ARN-Seq se estableció en paralelo al rápido desarrollo de una serie de tecnologías de secuenciación de ADN de alto rendimiento. Sin embargo, antes de la secuenciación de los transcritos de ARN extraídos, se realizan diferentes pasos clave de procesamiento. Los métodos difieren en el uso del enriquecimiento de los transcritos, fragmentación, amplificación, secuenciación simple o por pares, y en la conservación o no de la información de la hebra.
La sensibilidad de un experimento de ARN-Seq puede incrementarse al enriquecer tipos de ARN que sean de interés y eliminando los ARNs conocidos en abundancia. Las moléculas de ARNm se pueden separar utilizando sondas de oligonucleótidos, las cuales se unen a sus colas de poli-A. De manera alternativa, se puede utilizar la ribo-depleción para eliminar específicamente ARNr abundante y no informativo mediante hibridación de sondas a medida de las secuencias de ARNr específicas de taxón (ej.: ARNr de mamífero, de planta). Sin embargo, la ribo-depleción también puede introducir cierto sesgo al eliminar de manera inespecífica transcritos fuera del objetivo. Los ARNs pequeños, tales como micro ARNs, se pueden purificar en función de su tamaño mediante electroforesis en gel y extracción.
Dado que los ARNs mensajeros son más largos que las lecturas de los métodos de secuenciación de alto rendimiento típicos, los transcritos se suelen fragmentar antes de la secuenciación. El método de fragmentación es un aspecto clave de la construcción de bibliotecas para secuenciación. La fragmentación se puede lograr mediante hidrólisis química, nebulización, sonicación o transcripción inversa con nucleótidos terminadores de cadena. Alternativamente, la fragmentación y marcado de ADNc se pueden hacer simultáneamente mediante el uso de enzimas transposasas.
Durante la separación para la secuenciación, las copias ADNc de los transcritos se pueden amplificar mediante PCR para enriquecer los fragmentos que contienen las secuencias de los adaptadores 5' y 3'. La amplificación también se utiliza para permitir la secuenciación de cantidades muy pequeñas de ARN, siendo hasta 50 pq en aplicaciones extremas. Los controles del aumento de ARNs conocidos se pueden utilizar para controles de calidad de la preparación de bibliotecas y secuenciación, en términos de contenido en GC, longitud de los fragmentos, así como el sesgo producido por la posición de los fragmentos en un transcrito. Los identificadores moleculares únicos (UMIs) son secuencias cortas al azar que se utilizan para marcar individualmente fragmentos de secuencia durante la preparación de genotecas para que cada fragmento marcado sea único. Los UMIs proveen de una escala absoluta para la cuantificación, la oportunidad de corregir el consiguiente sesgo de amplificación introducido durante la construcción de la genoteca, y estimar de manera precisa el tamaño de las muestras iniciales. Los UMIs son particularmente adecuados para transcriptómica mediante ARN-Seq de célula única, donde la cantidad de ARN de entrada está restringida y se necesita la extensión de la amplificación de la muestra.
Una vez que se han preparado las moléculas de los transcritos, se pueden secuenciar en una sola dirección (single-end) o en ambas direcciones (paired-end). Una secuencia en una sola dirección suele ser más rápida de producir, mas barata que la secuenciación en ambas direcciones y suficiente para la cuantificación de niveles de expresión génica. La secuenciación en dos direcciones produce alineamientos/ensamblajes más robustos, lo cual es beneficioso para la anotación de genes y descubrimiento de isoformas de transcritos. Los métodos de ARN-Seq específicos de hebra preservan la información de la hebra de un transcrito secuenciado. Sin esta información, las lecturas se pueden alinear al locus de un gen pero no informan en qué dirección se transcribe el gen. Este tipo de ARN-Seq es útil para descifrar la transcripción de genes que se solapan en diferentes direcciones y para hacer predicciones de genes más robustas en organismos no modelo.
| Plataforma | Lanzamiento comercial | Longitud típica de lectura | Rendimiento máximo por análisis | Precisión de lectura única | Análisis de ARN-Seq depositadas en NCBI SRA (octubre de 2016) |
|---|---|---|---|---|---|
| 454 Life Sciences | 2005 | 700 pb | 0,7 Gbp | 99,9% | 3 548 |
| Illumina | 2006 | 50–300 pb | 900 Gbp | 99,9% | 362 903 |
| SOLiD | 2008 | 50 pb | 320 Gbp | 99,9% | 7 032 |
| Ion Torrent | 2010 | 400 pb | 30 Gbp | 98% | 1 953 |
| PacBio | 2011 | 10 000 pb | 2GB | 87% | 160 |
Leyenda: NCBI SRA - Archivo de lecturas de secuencias del Centro Nacional para la Información Biotecnológica (NCBI)
Actualmente, el ARN-Seq se basa en copiar moléculas de ARN en ADNc previamente a la secuenciación; por lo tanto, las plataformas consiguientes son las mismas para datos genómicos y transcriptómicos. Consecuentemente, el desarrollo de tecnología de secuenciación de ADN ha sido una característica definitoria del ARN-Seq. La secuenciación directa de ARN mediante secuenciación de nanoporos representa una técnica de ARN-Seq de vanguardia en la actualidad. La secuenciación de ARN con nanoporos puede detectar bases modificadas que pasarían inadvertidas de otra manera al secuenciar ADNc y también elimina pasos de amplificación que podrían introducir sesgos.
La sensibilidad y precisión de un experimento de ARN-Seq dependen del número de lecturas obtenidas por cada muestra. Se necesita una gran cantidad de lecturas para asegurar una cobertura suficiente del transcriptoma, permitiendo la detección de transcritos de baja abundancia. El diseño experimental se complica aún más por las tecnologías de secuenciación con un rango limitado de salida de resultados, la eficiencia variable de la creación de secuencias, y la calidad de secuencia variable. Además de estas consideraciones, en cada especie existe un número diferente de genes y, por lo tanto, requiere un rendimiento de secuencias adaptado para un transcriptoma eficaz. Estudios iniciales determinaron los umbrales adecuados empíricamente, pero a medida que la tecnología maduró se predijo computacionalmente la cobertura adecuada mediante la saturación del transcriptoma. Aunque es algo contraintuitivo, la manera más efectiva de mejorar la detección de expresión diferencial en genes de baja expresión es añadir más replicas biológicas en vez de añadir más lecturas. Los estándares actuales recomendados por el proyecto de la Enciclopedia de elementos de ADN (ENCODE) son una cobertura de 70 veces el exoma para el ARN-Seq estándar y de hasta 500 veces el exoma para detectar transcritos e isoformas raros.
Análisis de datos
Los métodos de transcriptómica son altamente paralelos y requieren una computación significativa para producir datos significativa tanto para experimentos de microarrays como ARN-Seq. Los datos de microarray se registran como imágenes de alta resolución, necesitando la detección de características y análisis espectral. Cada archivo de datos crudos de microarray tiene un tamaño de 750 MB aproximadamente, mientras que las intensidades procesadas, en torno a 60 MB. El acoplamiento de múltiples sondas pequeñas con un solo transcrito puede revelar detalles sobre la estructura intrón-exón, requiriendo de modelos estadísticos para determinar la autenticidad de la señal de los resultados. Los estudios de ARN-Seq producen miles de millones de secuencias cortas de ADN, las cuales se tienen que alinear a genomas de referencia compuestos de millones a miles de millones de pares de bases. El ensamblaje de novo de las lecturas en un conjunto de datos requiere la construcción de grafos de secuencias altamente complejos. Los protocolos de ARN-Seq son altamente repetitivos y se benefician de la computación paralela, pero los algoritmos modernos permiten que el hardware informático de consumo es suficiente para experimentos simples de transcriptómica que no requieren de ensamblaje de novo de las lecturas. Un transcriptoma humano podría ser capturado eficazmente utilizando ARN-Seq con 30 millones de secuencias de 100 pb por muestra. Este ejemplo requeriría de aproximadamente 1,8 GBs de espacio de disco por muestra al almacenar los datos en formato comprimido FASTQ. Los datos de conteo procesados para cada gen ocuparían mucho menos espacio, siendo equivalentes a las intensidades procesadas en un microarray. Los datos de secuenciación se pueden almacenar en repositorios públicos, tales como el Sequence Read Archive (SRA). Se pueden cargar conjuntos de datos de ARN-Seq mediante la plataforma Gene Expression Omnibus.
Procesamiento de imágenes

El procesamiento de imágenes de microarray debe identificar correctamente la cuadrícula regular de características en una imagen y cuantificar independientemente la intensidad de fluorescencia para cada característica. Adicionalmente, se deben identificar los artefactos en imágenes y eliminarlos del análisis general. Las intensidades de fluorescencia indican la abundancia de cada secuencia, ya que la secuencia de cada sonda en el array se conoce previamente.
Los primeros pasos del ARN-Seq también incluyen un procesado similar de imágenes; sin embargo, la conversión de imágenes a datos de secuencias se trata típicamente de manera automática mediante software. El método de secuenciación por síntesis de Illumina resulta en un array de clústeres distribuidos sobre la superficie de una celda de flujo. La celda de flujo se visualiza hasta cuatro veces durante cada ciclo de secuenciación, con un total de decenas a cientos de ciclos. Los clústers de celdas de flujo son análogos a los puntos de un microarray y se deben identificar correctamente durante los primeros estadíos del proceso de secuenciación. En el método de pirosecuenciación de Roche, la intensidad de luz emitida determina el número de nucleótidos consecutivos en una repetición homopolimérica. Existen muchas variantes de estos métodos, cada cual con un perfil de error diferente para los datos resultantes.
Análisis de datos de ARN-Seq
Los experimentos de ARN-Seq generan grandes cantidades de lecturas de secuencias en crudo que tienen que ser procesadas para obtener información útil. El análisis de datos normalmente requiere una combinación de herramientas de software bioinformático (ver también Anexo: Herramientas bioinformáticas de ARN-Seq) que varían en función del diseño experimental y objetivos. El proceso se puede dividir en cuatro etapas: control de calidad, alineamiento, cuantificación y expresión diferencial. Los programas más populares de ARN-Seq se ejecutan desde una interfaz de líneas de comandos, ya sea en un ambiente Unix o en un ambiente estadístico de R/Bioconductor.
Control de calidad
Las lecturas de secuenciación no son perfectas, por lo que la precisión de cada base en la secuencia necesita estimarse mediante posteriores análisis. Los datos crudos se examinan para asegurar que las puntuaciones de calidad de las llamadas de bases son altas, que el contenido en GC coincide con la distribución esperada, que los motivos de secuencias cortas (k-meros) no están sobrerrepresentados, y que el ratio de lecturas duplicadas es aceptablemente bajo. Existen diferentes opciones de software para en análisis de calidad de las secuencias, incluyendo FastQC y FaQCs. Las anormalidades se pueden eliminar o marcar para tratamientos especiales en procesos posteriores.
Alineamiento
Para relacionar la abundancia de lecturas de secuencias con la expresión de un gen en particular, se alinea las secuencias de transcritos a un genoma de referencia, o entre las secuencias en un ensamblaje de novo si no existe un genoma de referencia. Los retos clave para el software de alineamiento incluyen: suficiente velocidad para permitir el alineamiento de miles de millones de secuencias cortas, flexibilidad para reconocer y tratar el empalme de intrones de ARNm eucariótico, y la correcta asignación de lecturas que se mapean en diferentes localizaciones del genoma. Los avances en software han atajado enormemente estos problemas, y el incremento en la longitud de lecturas secuenciadas reduce la probabilidad de alineamientos ambiguos. El Instituto Europeo de Bioinformática (EBI) soporta actualmente una lista de alineadores de secuencias de alto rendimiento.
El alineamiento de secuencias de transcritos primarios derivados de eucariotas a un genoma de referencia requiere de un tratamiento especializado de las secuencias de intrones, los cuales no están presentes en el ARNm maduro. Los alineadores de lecturas cortas realizan una ronda adicional de alineamientos específicamente diseñados para identificar las uniones de empalme, basándose en las secuencias canónicas de los sitios de empalme y la información conocida de sitios de empalme de intrones. La identificación de los sitios de empalme de intrones previene el alineamiento incorrecto de estos o de ser erróneamente descartados, permitiendo el alineamiento de más lecturas al genoma de referencia y la mejora de la precisión en las estimaciones de expresión génica. Debido a que la regulación génica puede ocurrir a nivel de isoformas de ARNm, los alineamientos en los que se tenga en cuenta el empalme alternativo también permiten la detección de cambios en la abundancia de isoformas, cuya información se perdería en otros análisis en bloque.
El ensamblaje de novo se puede utilizar para alinear lecturas entre sí para construir secuencias de transcritos en su longitud completa sin usar un genoma de referencia. Los retos particulares de un ensamblaje de novo incluyen requerimiento computacionales mayores comparados con un transcriptoma basado en una referencia, validación adicional de variantes génica o fragmentos, y anotación funcional de transcritos ensamblados. Las primeras métricas utilizadas para describir ensamblajes de transcriptoma, tales como N50, han demostrado ser engañosas y ahora se dispone de métodos de evaluación mejorados. Las métricas basadas en anotación evalúan mejor cómo de completo es el ensamblaje, tales como el conteo de mejores cóntigos recíprocos. Una vez se ha completado el ensamblaje de novo, este se puede usar como referencia para métodos posteriores de alineamiento de secuencias y para análisis cuantitativo de expresión génica.
| Software | Año de lanzamiento | Última actualización | Eficiencia computacional | Fortalezas y debilidades |
|---|---|---|---|---|
| Velvet-Oases | 2008 | 2011 | Baja, un solo subproceso, necesita RAM alta | El ensamblador original de lecturas cortas. Ahora está en gran parte reemplazado. |
| SOAPdenovo-trans | 2011 | 2014 | Moderada, múltiples subprocesos, RAM media | Un ejemplo temprano de un ensamblador de lecturas cortas. Se ha actualizado para el ensamblaje del transcriptoma. |
| Trans-ABySS | 2010 | 2016 | Moderada, múltiples subprocesos, RAM media | Adecuado para lecturas cortas, puede manejar transcriptomas complejos y está disponible una versión paralela de MPI para clústeres informáticos. |
| Trinity | 2011 | 2017 | Moderada, múltiples subprocesos, RAM media | Adecuado para lecturas cortas. Puede manejar transcriptomas complejos pero requiere mucha memoria. |
| miraEST | 1999 | 2016 | Moderada, múltiples subprocesos, RAM media | Puede procesar secuencias repetitivas, combinar diferentes formatos de secuenciación y se acepta una amplia gama de plataformas de secuencias. |
| Newbler | 2004 | 2012 | Baja, un único subproceso, RAM alta | Especializado para adaptarse a los errores de secuenciación de homopolímeros típicos de los secuenciadores Roche 454. |
| CLC genomics workbench | 2008 | 2014 | Alta, múltiples subprocesos, RAM baja | Tiene una interfaz gráfica de usuario, puede combinar diversas tecnologías de secuenciación, no tiene características específicas del transcriptoma y se debe comprar una licencia antes de su uso. |
| SPAdes | 2012 | 2017 | Alta, múltiples subprocesos, RAM baja | Se utiliza para experimentos de transcriptómica en células individuales. |
| RSEM | 2011 | 2017 | Alta, múltiples subprocesos, RAM baja | Puede estimar la frecuencia de transcritos con empalme alternativo. Fácil de usar. |
| StringTie | 2015 | 2019 | Alta, múltiples subprocesos, RAM baja | Puede usar una combinación de métodos de ensamblaje guiados por un genoma de referencia y de novo para identificar transcritos. |
Leyenda: RAM – memoria de acceso aleatorio; MPI: interfaz de paso de mensajes; EST: marcador de secuencia expresada.
Cuantificación
La cuantificación de alineamientos de secuencias se puede realizar a nivel de gen, exón o de transcrito. Los resultados típicos incluyen una tabla de conteo de lecturas para cada característica analizada por el software; por ejemplo, para genes en un archivo de formato general feature. El conteo de genes y exones se puede calcular fácilmente utilizando HTSeq, por ejemplo, La cuantificación a nivel de transcrito es más complicada y requiere de métodos probabilísticos para estimar la abundancia de isoformas de transcritos a partir de lecturas cortas; por ejemplo, utilizando el software cufflinks. Las lecturas alineadas equitativamente a múltiples localizaciones se deben identificar y eliminar, alinear a una de las posibles localizaciones, o alinear a la localización más probable.
Algunos métodos de cuantificación pueden eludir por completo la necesidad de un alineamiento exacto de una lectura a un genoma de referencia. El software kallisto es un método que combina el pseudoalineamiento y cuantificación en un solo paso, ejecutándose 2 ordenes de magnitud más rápido que otros métodos contemporáneos, tales como aquellos utilizados por tophat/cufflinks, con menos carga computacional.
Expresión diferencial
Una vez que el conteo de cada transcrito está disponible, la expresión génica diferencial se mide mediante normalización, modelización, y análisis estadístico de los datos. La mayoría de las herramientas leen una tabla de genes y su conteo, pero algunos problemas, como cuffdiff, aceptan alineamiento de lecturas en formato BAM. Los resultados finales de estos análisis son listas de genes con tests por pares asociados para expresión diferencial entre tratamientos y las estimaciones de probabilidad de esas diferencias.
| Software | Ambiente | Especialización |
|---|---|---|
| Cuffdiff2 | Basado en Unix | Análisis de transcrito que rastrea empalmes alternativos de ARNm |
| EdgeR | R/Bioconductor | Cualquier dato genómico basado en conteo |
| DEseq2 | R/Bioconductor | Tipos de datos flexibles, baja replicación |
| Lima/Voom | R/Bioconductor | Datos de micromatrices o ARN-Seq, diseño de experimentos flexible |
| Ballgown | R/Bioconductor | Descubrimiento de transcritos eficiente y sensible, flexible. |
Leyenda: ARNm - ARN mensajero.
Validación
Los análisis de transcriptómica se pueden validar utilizando una técnica independiente, por ejemplo, una PCR cuantitativa (qPCR), la cual es reconocible y estadísticamente evaluable. La expresión génica se mide en comparación con estándares definidos tanto para el gen de interés como para genes control. La medición por qPCR es similar a la obtenida por ARN-Seq, en la que se puede calcular un valor para la concentración de una región diana en una muestra dada. Sin embargo, la qPCR está restringida por amplicones más pequeños de 300 pb, normalmente hacia el extremo 3' de la región codificante, evitando la región 3'-UTR. Si se necesita la validación de isoformas de transcritos, una inspección de los alineamiento de lecturas en ARN-Seq debería indicar donde se podrían localizar los primers de la qPCR para una discriminación máxima. La medición de múltiples genes control con los genes de interés genera una referencia estable en un contexto biológico. La validación de datos de ARN-Seq mediante qPCR ha demostrado normalmente que los diferentes métodos de ARN-Seq están altamente correlacionados.
La validación funcional de genes clave es una consideración importante para la planificación posterior a la construcción del transcriptoma. Los patrones de expresión génica observados se pueden relacionar funcionalmente a un fenotipo mediante un estudio independiente de knockdown/rescate en el organismo de interés.
Aplicaciones
Diagnóstico y perfilado de enfermedades
Las estrategias de transcriptómica han visto una amplía aplicación en diferentes áreas de la investigación biomédica, incluyendo diagnóstico y clasificación de enfermedades. Las aproximaciones de ARN-Seq han permitido la identicación a gran escala de sitios de inicio de la transcripción, descubierto el uso de promotores alternativos y nuevas variantes de empalme. Estos elementos reguladores son importantes en enfermedades humanas y, por lo tanto, definir tales variantes es crucial para la interpretación de estudios de asociación de enfermedades. El ARN-Seq puede identificar también polimorfismos de nucleótido único (SNPs) asociados a enfermedades, expresión específica de alelo, y fusiones de genes, los cuales contribuyen al entendimiento de variantes causales de enfermedad.
Los retrotransposones son elementos transponibles que proliferan en genomas eucariotas mediante un proceso que involucra la transcripción inversa. El ARN-Seq puede proveer de información sobre la transcripción de retrotransposones endógenos que pueden influenciar la transcripción de genes vecinos por diferentes mecanismos epigenéticos que llevan a enfermedad. De manera similar, el potencial de utilizar ARN-Seq para comprender enfermedades relacionados con el sistema inmunitario se está expandiendo rápidamente debido a la habilidad de diseccionar poblaciones de células inmunitarias y secuenciar repertorios de receptores de células T y B de pacientes.
Transcriptomas humanos y patógenos
El ARN-Seq de patógenos humanos se ha convertido en un método establecido para cuantificar cambios en la expresión génica, identificando nuevos factores de virulencia, prediciendo resistencia a antibióticos, y desentrañando interacciones inmunitarias huésped-patógeno. Un objetivo principal de esta tecnología es desarrollar mediciones de control de infección optimizadas y tratamientos individualizados dirigidos.
El análisis transcriptómico se ha centrado predominantemente en ya sea el huésped o el patógeno. El ARN-Seq dual se ha aplicado para clasificar simultáneamente la expresión de ARN tanto en el patógeno como en el huésped a lo largo del proceso de infección. Esta técnica permite estudiar la respuesta dinámica y redes reguladoras de genes inter-especie en ambas partes involucradas en la interacción desde el contacto inicial hasta la invasión y la persistencia final del patógeno o su eliminación por el sistema inmunitario del huésped.
Respuestas al entorno
La transcriptómica permite la identificación de genes y rutas metabólicas que responden a y contrarrestan el estrés ambiental biótico y abiótico. La naturaleza no dirigida de la transcriptómica permite la identificación de redes transcripcionales nuevas en sistemas complejos. Por ejemplo, el análisis comparativo de una serie de líneas de garbanzo en distintas fases de desarrollo identificó perfiles transcripcionales distintos asociados al estrés de sequía y salinidad, incluida la identificación del rol de isoformas de transcritos de AP2-EREBP. La investigación de la expresión génica durante la formación de biopelículas por el hongo patógeno Candida albicans reveló un conjunto de genes co-regulados, los cuales son críticos para el establecimiento de la biopelícula y su mantenimiento.
Los perfiles transcriptómicos también aportan información crucial sobre los mecanismos de resistencia a fármacos. Un análisis de más de 1000 aislados de Plasmodium falciparum, un parásito virulento responsable de la malaria en humanos, identificó que la regulación al alza de la respuesta a las proteínas desplegadas y que una progresión más lenta durante los estadios tempranos del ciclo de desarrollo asexual intraeritrocítico se asociaban con la resistencia a la artemisinina en aislados del sudeste asiático.
El uso de la transcriptómica también es importante para investigar respuestas en el ambiente marino. En ecología marina, "estrés" y "adaptación" han figurado entre los temas de investigación más comunes, especialmente en relación con el estrés antropogénico, como el cambio global o la contaminación. La mayoría de los estudios en este campo se han realizado en animales, aunque los invertebrados han estado infrarrepresentados. Un problema que sigue existiendo es la deficiencia de estudios de genética funcional, que dificulta la anotación de genes, especialmente para especies no modelo, y puede llevar a conclusiones vagas sobre los efectos a las respuestas estudiadas.
Anotación funcional de genes
Todas las técnicas transcriptómicas han sido particularmente útiles para identificar las funciones de genes e identificar aquellos responsables de determinados fenotipos. La transcriptómica de ecotipos de Arabidopsis que hiperacumulan metales correlacionó genes involucrados en la absorción, tolerancia y homeostasis de metales con el fenotipo. La integración de conjuntos de datos de ARN-Seq en diferentes tejidos se ha utilizado para mejorar la anotación funcional de genes en organismos de importancia comercial (ej.: pepino) o especies en peligro (ej.: koala).
El ensamblaje de lecturas de ARN-Seq no depende de un genoma de referencia y, por ello, es ideal para estudios de la expresión génica de organismos no modelo con recursos genómicos inexistentes o pobremente desarrollados. Por ejemplo, una base de datos de SNPs utilizada en programas de mejora genética del abeto Douglas se creó mediante análisis transcriptómico de novo en ausencia de un genoma secuenciado. De manera similar, se identificaron los genes involucrados en el desarrollo de tejidos cardiaco, muscular y nervioso en langostas mediante la comparación de transcriptomas de varios tipos de tejidos sin el uso de la secuencia de un genoma. El ARN-Seq también se puede utilizar para identificar regiones codificantes de proteínas previamente desconocidas en genomas ya secuenciados.
Un reloj del envejecimiento basado en transcriptomas
Las intervenciones preventivas del envejecimiento no son posibles sin mediciones de la velocidad de envejecimiento personal. La forma más actualizada y compleja de medir el envejecimiento es mediante el uso de biomarcadores variables del envejecimiento humano, lo cual se basa en la utilización de redes neuronales profundas que se pueden entrenar con cualquier tipo de datos ómicos para predecir la edad del sujeto. Se ha demostrado que el envejecimiento es un fuerte impulsor de cambios en el transcriptoma. Los relojes de envejecimiento basados en transcriptomas han sufrido de considerables variaciones en los datos y una precisión relativamente baja. Sin embargo, una aproximación que utiliza el escalado temporal y binarización de transcriptomas para definir un conjunto de genes que predice la edad biológica con precisión permitió alcanzar una evaluación cercana al límite teórico.
ARN no codificante
La transcriptómica se aplican más comúnmente al contenido en ARNm de una células. Sin embargo, las mismas técnicas son igualmente aplicables a ARNs no codificantes (ARNnc), los cuales no se traducen a proteínas, pero en cambio tienen funciones directas (ej.: papel en la traducción de proteínas, replicación del ADN, empalme de ARN, y regulación transcripcional). Muchos de estos ARNnc afectan al estado de enfermedades, incluyendo el cáncer, enfermedades cardiovasculares y neurológicas.
Bases de datos de transcriptómica
Los estudios de transcriptómica generan grandes cantidades de datos que tienen potenciales aplicaciones más allá de los objetivos originales de un experimento. Como tales, los datos crudos o procesados se pueden depositar en bases de datos públicas para asegurar su utilidad para la comunidad científica en general. Por ejemplo, a fecha de 2018, la base de datos Gene Expression Omnibus contenía millones de experimentos.
| Nombre | Anfitrión | Datos | Descripción |
|---|---|---|---|
| Gene Expression Omnibus | NCBI | Microarray ARN-Seq | Primera base de datos transcriptómica que acepta datos de cualquier fuente. Introdujo los estándares comunitarios MIAME y MINSEQE que definen los metadatos experimentales necesarios para garantizar una interpretación y repetibilidad efectivas. |
| ArrayExpress | ENA | Microarray | Importa conjuntos de datos de Gene Expression Omnibus y acepta envíos directos. Los datos procesados y los metadatos del experimento se almacenan en ArrayExpress, mientras que las lecturas de secuencia sin procesar se mantienen en ENA. Cumple con los estándares de MIAME y MINSEQE. |
| Expression Atlas | EBI | Microarray ARN-Seq | Base de datos de expresión génica específica de tejido para animales y plantas. Muestra análisis secundarios y visualización, como el enriquecimiento funcional de términos de ontología génica, dominios InterPro o rutas metabólicas. Enlaces a datos de abundancia de proteínas donde estén disponibles. |
| Genevestigator | Datos procesados privadamente | Microarray ARN-Seq | Contiene procesamientos manuales de conjuntos de datos de transcriptomas públicos, centrándose en datos médicos y de biología vegetal. Los experimentos individuales se normalizan en la base de datos completa para permitir la comparación de la expresión génica en diversos experimentos. La funcionalidad completa requiere la compra de una licencia, con acceso gratuito a una funcionalidad limitada. |
| RefEx | DDBJ | Todos | Transcriptomas humanos, murinos y de rata de 40 órganos diferentes. Expresión génica visualizada como mapas de calor proyectados en representaciones 3D de estructuras anatómicas. |
| NONCODE | nocode.org | ARN-Seq | ARN no codificantes (ARNnc) excluyendo ARNt y ARNr. |
Leyenda: NCBI – Centro Nacional para la Información Biotecnológica; EBI – Instituto Europeo de Bioinformática; DDBJ – Banco de datos de ADN de Japón; ENA – Archivo Europeo de Nucleótidos; MIAME – Información Mínima Sobre un Experimento de Microarray; MINSEQE – Información mínima sobre un experimento de secuenciación de nucleótidos de alto rendimiento.
Véase también
Notas
Otras lecturas
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. (Mayo de 2017). «Transcriptomics technologies». PLOS Computational Biology 13 (5): e1005457. Bibcode:2017PLSCB..13E5457L. PMC 5436640. PMID 28545146. doi:10.1371/journal.pcbi.1005457.
- Taguchi, Y. H. (2019). «Comparative Transcriptomics Analysis». Encyclopedia of Bioinformatics and Computational Biology: 814-818. ISBN 9780128114322. doi:10.1016/B978-0-12-809633-8.20163-5.
- Software utilizado en transcriptómica: