
En química orgánica, la síntesis de péptidos es la producción de péptidos, compuestos orgánicos en los que numerosos aminoácidos se encuentran unidos mediante enlaces peptídicos, llamados también enlaces amida. El proceso biológico de producción de polipéptidos (proteínas) se conoce como biosíntesis de proteínas.

Química
La síntesis peptídica tiene lugar cuando el grupo carboxilo (C-terminal) de un aminoácido se une al grupo amino (N-terminal) de otro. Normalmente, debido a la posibilidad de reacciones no deseadas, la presencia de grupos protectores se hace necesaria. La reacción química de síntesis de un péptido empieza en el extremo C-terminal del péptido y acaba en su extremo N-terminal. En esto difiere de la biosíntesis proteica, que comienza en el extremo N-terminal.
Síntesis en solución
La síntesis en solución o fase líquida, el método clásico en la síntesis de péptidos, ha sido reemplazada en la mayoría de laboratorios por la síntesis en fase sólida (ver más adelante). Pese a ello, no deja de ser útil cuando es preciso producir péptidos a gran escala, con fines industriales.
Síntesis en fase sólida (SPPS)
La síntesis peptídica en fase sólida (SPPS en sus siglas en inglés), cuyo pionero fue Robert Bruce Merrifield, constituyó un cambio de paradigma para la comunidad científica dedicada a la síntesis química de péptidos. En la actualidad es el método usado en la creación de péptidos y proteínas en laboratorio. La SPPS permite sintetizar péptidos naturales difíciles de expresar en bacterias, incorporar aminoácidos no proteicos, modificar el esqueleto de péptidos y proteínas y sintetizar proteínas a partir de D-aminoácidos.
En esta síntesis, pequeñas bolas sólidas, porosas pero insolubles, son tratadas con unidades funcionales (agentes de acoplamiento) sobre las que se construyen las cadenas peptídicas. El péptido permanece unido covalentemente a la bola hasta que es desanclado por reactivos como el fluoruro de hidrógeno líquido o el ácido trifluoroacético (TFA). El péptido queda así inmovilizado en la fase sólida y permanece retenido durante el proceso de filtrado, mientras que los reactivos de la fase líquida y los subproductos de la síntesis son eliminados.
La SPPS sigue una pauta general de ciclos repetitivos de acoplamiento-lavado-desprotección-lavado. El extremo libre amino terminal de un péptido unido en una fase sólida se acopla (ver más adelante) a una única unidad aminoacídica N protegida. Esta unidad es entonces desprotegida, mostrando así un nuevo extremo amino al cual puede unirse otro aminoácido. La superioridad de esta técnica reside en parte en la habilidad con que se lleven a cabo los ciclos de lavado después de cada reacción, en los que se elimina el exceso de reactivo mientras que el péptido de interés permanece unido covalentemente a la resina insoluble.
La cuestión más importante aquí es obtener en cada etapa un rendimiento lo más elevado posible. Por ejemplo, si se pretende un rendimiento del 99% en cada ciclo de acoplamiento, un péptido de 26 aminoácidos acaba sintetizado con un rendimiento neto del 77% (asumiendo un rendimiento del 100% en cada desprotección); si se espera un rendimiento del 95% en cada ciclo, el péptido es sintetizado con un rendimiento final del 25% . Para ello cada aminoácido es añadido en exceso y el acoplamiento entre aminoácidos se optimiza gracias a una serie de reactivos determinados.
Los dos métodos de síntesis peptídica más habitualmente usados son el Fmoc y el t-Boc. Al contrario que en la síntesis proteica llevada a cabo por los ribosomas, la síntesis peptídica en fase sólida progresa desde el extremo C-terminal al extremo N-terminal. Los N-términos de los monómeros aminoacídicos son protegidos por estos dos grupos y añadidos a una cadena aminoacídica desprotegida.
Si bien se dispone de sintetizadores automatizados que realizan ambas técnicas, aún hoy muchos equipos de investigación continúan realizando la SPPS manualmente.
La SPPS presenta limitaciones de rendimiento (este disminuye a medida que las reacciones se suceden) y normalmente los péptidos y proteínas de alrededor de 70 aminoácidos constituyen el límite de las posibilidades de la polimerización sintética. Estas limitaciones tienen que ver también con la secuencia, siendo los péptidos y las proteínas amiloides normalmente difíciles de obtener. Sin embargo, pueden conseguirse polímeros de mayor longitud usando el ligamiento químico nativo, con un rendimiento cuantitativo.
Desde que se introdujo hace más de 40 años, la SPPS ha mejorado sustancialmente. Para empezar, se han optimizado las resinas originales. Además, se ha logrado que los agentes de acoplamiento que actúan entre el aminoácido C-terminal y la resina de poliestireno aumenten la cantidad de producto obtenido mediante la mejora de los procesos de unión y desanclaje. Por otro lado, los grupos que protegen las cadenas laterales han evolucionado reduciendo la aparición de reacciones colaterales no deseadas. A esto hay que añadir la introducción de nuevos grupos activadores del extremo carboxilo del aminoácido añadido, que han optimizado el acoplamiento y han reducido la epimerización. En general se ha mejorado todo el proceso en sí.
En el informe inicial de Merrifield, la desprotección del grupo amino alfa resultaba en la formación de una sal a partir del péptido y la resina, que requería ser neutralizada con una base antes del acoplamiento. En el tiempo que pasaba entre la neutralización del grupo amino y el acoplamiento con el siguiente aminoácido se producía la agregación entre péptidos, principalmente mediante la formación de estructuras secundarias, lo que afectaba negativamente al acoplamiento. El equipo de Kent demostró que si la neutralización se producía paralelamente a la unión del siguiente aminoácido, el acoplamiento mejoraba. Cada uno de estos progresos ha contribuido a que la SPPS sea en la actualidad un método robusto.
Síntesis peptídica Fmoc en fase sólida
La capacidad del fluoruro de hidrógeno para degradar las proteínas en las condiciones del desanclaje final condujo a la introducción de un nuevo protector del grupo amino alfa, basado en el 9-fluorenilmetoxicarbonilo (Fmoc). El método Fmoc permite una acción de desprotección más suave. Este método utiliza una base, normalmente la piperidina (20%-50%) en DMF para la eliminación del grupo Fmoc y que el grupo amino alfa quede expuesto y listo para reaccionar con un nuevo aminoácido activo. Al contrario que los métodos t-Boc, en los que se utiliza un ácido para desproteger el grupo amino alfa, la SPPS Fmoc usa una base, con lo cual la amina expuesta resulta neutra. Así pues, no es necesaria la posterior neutralización de la resina-péptido, si bien la ausencia de interacción electrostática de tipo repulsivo entre los péptidos puede aumentar el proceso de agregación.
Paralelamente al desarrollo del método Fmoc para la SPPS, se han creado diversas resinas que pueden ser retiradas por el TFA. De forma similar a la estrategia t-Boc, se usan dos resinas primarias, dependiendo de si se precisa una amida o un ácido carboxílico en el extremo C-terminal. La resina de Wang es la más usada para péptidos con ácidos carboxílicos C-terminales. Si se desea una amida C-terminal, entonces se usa la resina de Rink.
Los grupos protectores temporales se basan en el terc-butilo. El desanclaje final de la proteína de la resina y la posterior eliminación de los grupos protectores permanentes son llevadas a cabo por el TFA en presencia de recolectores. De este modo, el método Fmoc es ortogonal en dos direcciones: la desprotección del grupo amino alfa y el desanclaje final de la resina son mecanismos independientes. El producto final es una sal de TFA, más difícil de solubilizar que las sales de fluoruro generadas por la SPPS con el método t-Boc. Este método, por tanto, es más suave que el t-Boc, en la medida en que los ciclos de desprotección y desanclaje de la resina ocurren en otras condiciones, y no porque la velocidad de las reacciones sea diferente.
Síntesis peptídica t-Boc en fase sólida
El método original para la síntesis de proteínas se basaba en el terc-Butiloxicarbonilo (t-Boc) como protector temporal del grupo amino alfa. En este método, el grupo t-Boc se une covalentemente al grupo amino para eliminar su reactividad nucleofílica. El aminoácido C-terminal queda unido covalentemente a la resina a través de un agente de acoplamiento. A continuación, el grupo t-Boc es eliminado con un ácido como el TFA. Esto resulta en la formación de un grupo amino cargado positivamente, que queda simultáneamente neutralizado y acoplado a un aminoácido activo.
La compleción de las reacciones se ve facilitada por la adición en exceso del reactivo (del doble al cuádruple). Tras cada etapa de desprotección y acoplamiento se realiza un lavado con Dimetilformamida (DMF) para eliminar los reactivos remanentes, con lo que se consigue un elevado rendimiento (99%) en cada ciclo.
Cabe notar que las resinas se han mejorado mucho, lo que ha incrementado su capacidad para resistir el uso repetido de TFA en cada etapa de desprotección. Además, existen diferentes resinas que permiten el uso de distintos grupos en el extremo C-terminal. La resina de Fenilacetamidometilo (PAM) produce el clásico ácido carboxílico en posición C-terminal. Por su parte, la 4-Metilbenzhidrilamina (MBHA) produce una amida C-terminal, útil si se quiere mimetizar el interior de una proteína.
Normalmente se usa como protectores permanentes de las cadenas laterales el bencilo o los grupos basados en el bencilo. El desanclaje final del péptido transcurre en paralelo a la desprotección de la cadena lateral mediante fluoruro de hidrógeno líquido vía desanclaje hidrolítico. El producto resultante es una sal de fluoruro que puede solubilizarse fácilmente. Cabe recalcar que recolectores como el cresol se añaden al fluoruro de hidrógeno para evitar que los cationes reactivos terc-butilo generen productos no deseados. De hecho, el fluoruro de hidrógeno es muy fuerte y su uso puede degradar algunos péptidos, cosa que en su momento alentó el desarrollo de una técnica de SPPS más suave, basada en bases lábiles, como es el método Fmoc.
Algunos investigadores prefieren el método t-Boc para las síntesis complejas. Por otro lado, si lo que se quiere es producir péptidos sintéticos sensibles a la alcalinidad (como los depsipéptidos), se necesita un grupo protector t-Boc, ya que el método Fmoc usa una base para proteger el grupo amino alfa.
Comparativa de la síntesis peptídica Fmoc y t-Boc en fase sólida
Ambos métodos, el Fmoc y el t-Boc, ofrecen ventajas y desventajas. La elección de una u otra técnica dependerá de cada caso concreto.
| Boc | Fmoc | |
|---|---|---|
| Requiere de equipamiento especial | Sí | No |
| Coste de los reactivos | Bajo | Alto |
| Solubilidad de los péptidos | Mayor | Menor |
| Pureza de los péptidos hidrofóbicos | Elevada | Puede ser menor |
| Problemas de agregación | Poco frecuentes | Más frecuentes |
| Tiempo de síntesis | 20 min/aa | 20-60 min/aa |
| Desprotección final | HF | TFA |
| Seguridad | Potencialmente peligrosa | Relativamente segura |
| Ortogonal | No | Sí |
Con la SPPS t-Boc debe usarse un equipamiento especial para manejar la etapa de desprotección y desanclaje final, que requiere fluoruro de hidrógeno. Como el desanclaje final del péptido en el método Fmoc se realiza mediante TFA, este requerimiento no es necesario en esta técnica.
La solubilidad de los péptidos generados con la SPPS t-Boc es, por lo general, mayor que en aquellos sintetizados con el método Fmoc, ya que las sales de fluoruro son más solubles que las de TFA. A esto hemos de añadir los problemas de agregación que suelen darse en la SPPS Fmoc. Esto ocurre básicamente porque la eliminación del grupo t-Boc con TFA produce un grupo amino alfa cargado positivamente, mientras que la eliminación del grupo Fmoc da como resultado un grupo amino alfa neutro. Los impedimentos estéricos del grupo amino con carga positiva limitan la formación de la estructura secundaria sobre la resina. Por último, el método Fmoc se considera ortogonal en tanto en cuanto la desprotección del grupo amino se produce con una base, mientras que el desanclaje final de la resina tiene lugar con un ácido. Por el contrario, el método t-Boc utiliza un ácido para ambos procesos. Según esta comparativa, ambas técnicas poseen ventajas y desventajas, por lo que deben considerarse diversos factores a la hora de decidirse por una de ellas.
SPPS BOP
El uso del reactivo BOP fue descrito por primera vez por Castro et al., en 1975.
Soportes sólidos
El término “soporte sólido” parece indicar que las reacciones tienen lugar en la superficie de un soporte, aunque esto no es así en realidad: las reacciones se producen también en las partículas, por lo que la etiqueta “soporte sólido” describe más bien la insolubilidad del polímero.
Las propiedades físicas del soporte sólido, así como las aplicaciones que se le pueden dar, varían según el material con el que está hecho, la cantidad de entrecruzamientos que posea y el agente de acoplamiento que se use.
La mayoría de científicos de esta disciplina están de acuerdo en que el soporte debe tener una mínima cantidad de entrecruzamientos para conferirle estabilidad, lo que resulta en un sistema óptimamente sulfatado donde puede llevarse a cabo la síntesis peptídica en fase sólida. Las características de un soporte sólido eficaz son:
- Ser físicamente estable y permitir la filtración rápida de líquidos, como por ejemplo el exceso de reactivos.
- Ser inerte a todos los reactivos y disolventes usados en la SPPS.
- Tener buena capacidad de expansión en los disolventes para facilitar la penetración de los reactivos.
- Permitir la unión del primer aminoácido.
Hay cuatro grandes clases de soportes sólidos:
- Soporte sólidos de tipo gel: se trata de polímeros muy sulfatados con una distribución regular de grupos funcionales. Este tipo de soporte es el más común e incluye:
- Poliestireno: estireno entrecruzado con 1-2% de divinilbenceno.
- Poliacrilamida: la alternativa hidrofílica al poliestireno.
- Polietilenglicol (PEG): el PEG-poliestireno (PEG-PS) es más estable que el poliestireno y espacia los lugares de síntesis en el esqueleto del polímero.
- Soportes basados en PEG: compuestos por una red de polipropilenglicol-PEG o por PEG con poliamida o poliestrineo.
- 2.Soportes de tipo superficie: se han desarrollado muchos materiales para la funcionalidad de superficie, entre otros vidrio poroso controlado (CPG), fibras de celulosa y poliestireno con un alto grado de entrecruzamiento.
- Composites: polímeros de tipo gel soportados por matrices rígidas.
Resina de poliestireno
La resina de poliestireno es una resina versátil muy útil en la síntesis automatizada de péptidos debido a su limitada expansión en diclorometano. El soporte original usado por R. Bruce Merrifield era de poliestireno entrecruzado con 2% de divinilbenceno, soporte que se conoce como resina de Merrifield. Este soporte produce una bola hidrofóbica que es sulfatada por disolventes apolares como el diclorometano. A partir de ahí, se han ido desarrollando nuevas resinas con ventajas como una expansión o rigidez optimizadas (propiedad esta última que confiere robustez mecánica); o inercia química. Se ha desarrollado también un poliestireno con alto grado de entrecruzamiento (50%) que posee propiedades como una mayor estabilidad mecánica, un mejor grado de filtración de reactivos y disolventes y una rápida cinética de reacción.
Resina de poliamida
La resina de poliamida es también una resina útil y versátil. Parece hincharse un poco más que el poliestireno, por lo que puede no ser adecuada para su uso en sintetizadores automatizados con pocillos demasiado pequeños.
Resina híbrida de poliestireno y PEG (PEG-PS)
Un ejemplo de este tipo de resina es la resina Tentagel. La resina de base es el poliestireno sobre la que se unen largas cadenas (3000 Da) de polietilenglicol (PEG). La síntesis se lleva a cabo en el extremo distal del espaciador PEG, lo que la hace adecuada para péptidos complejos de gran longitud. Además, es también válida para la síntesis de bibliotecas combinatoriales de péptidos y para experimentos de screening de resinas. No se hincha mucho durante la síntesis lo que la convierte en la resina de elección en la síntesis peptídica robotizada.
Resina basada en PEG
ChemMatrix(R) es un nuevo tipo de resina entrecruzada basada en PEG, muy termoestable y quimioestable (compatible por tanto con la síntesis con microondas) y que ha mostrado, frente a las resinas basadas en el poliestireno, un mayor grado de expansión en acetonitrilo, diclorometano, DMF, N-metilpirrolidona, TFA y agua. Se ha comprobado que ChemMatrix(R) mejora sustancialmente la síntesis de secuencias hidrofóbicas y se recomienda en la síntesis de péptidos complejos de gran longitud.
Grupos protectores
Debido al exceso de aminoácidos que se utiliza para asegurar el acoplamiento completo durante cada etapa de la síntesis, se produce a menudo la polimerización en las reacciones en que los aminoácidos no están protegidos. Para evitar este fenómeno se usan grupos protectores o bloqueadores, lo que supone pasos de desprotección adicionales en la reacción sintética, creándose un patrón repetitivo como sigue:
- El grupo protector se retira de la cadena aminoacídica en una reacción de desprotección.
- Los reactivos son eliminados con el fin de crear un ambiente óptimo para el acoplamiento.
- Los aminoácidos protegidos disueltos en disolventes como la dimetilformamida combinados con agentes de acoplamiento son empujados en la columna de síntesis.
- Los agentes de acoplamiento son eliminados para crear un ambiente limpio para la desprotección.
En la actualidad, los grupos protectores que se usan normalmente en la SPPS son dos, el t-Boc y el Fmoc. Su labilidad se debe al grupo carbamato, que libera CO2 en el paso irreversible de desacoplamiento.
Grupo t-Boc
El grupo t-Boc (terc-Butoxicarbonilo o simplemente Boc) se usaba normalmente para bloquear el extremo amina del péptido, lo que requería el uso de grupos ácidos más estables para la protección de las cadenas laterales en las estrategias ortogonales. Se considera útil todavía porque disminuye el grado de agregación de los péptidos durante la síntesis. Los grupos t-Boc se unen a los aminácidos con un anhidrido y una base adecuada.
Grupo Fmoc

El Fmoc (9-fluorenilmetoxicarbonilo) es un grupo protector de amplio uso hoy en día, que se elimina normalmente del extremo N-terminal del péptido. La ventaja del Fmoc es que puede desanclarse en condiciones alcalinas muy suaves (por ejemplo en piperidina) y es estable en condiciones ácidas, aunque esto puede no cumplirse en determinados casos. Esto permite el uso de grupos protectores lábiles ligeramente ácidos, estables en álcali (como son el t-Boc y los grupos bencilo) para bloquear las cadenas laterales de los residuos aminoacídicos del péptido. Esta estrategia ortogonal es común en el arte de sintetizar péptidos orgánicos.
Por lo general, suele preferirse el Fmoc al t-Boc debido a la facilidad con que permite el desanclaje. Sin embargo, el grupo fluorenilo es mucho más grande que el grupo terc-butilo. En consecuencia, el coste de los aminoácidos con el método Fmoc se mantuvo elevado hasta la obtención a gran escala, en los años 90, de la enfuvirtida, uno de los primeros fármacos polipeptídicos. A partir de ese momento la demanda del mercado ajustó el precio de ambos tipos de aminoácidos.
Como el grupo fluorenilo liberado es un cromóforo, la desprotección por Fmoc puede monitorizarse mediante la lectura de la absorbancia del flujo con un detector ultravioleta, estrategia empleada en los sintetizadores automatizados.
Grupo Benzoxicarbonilo
Max Bergmann fue el primero en usar un grupo Z para sintetizar oligopéptidos. Otro grupo basado en el carbamato es el grupo benzoxicarbonilo, que se retira en condiciones menos suaves (en HBr o ácido acético) o por hidrogenación catalítica. Hoy se utiliza casi exclusivamente para la protección de las cadenas laterales.
Grupo Alloc
El grupo protector aliloxicarbonilo (Alloc o Aloc) se usa frecuentemente para proteger un ácido carboxílico, un hidroxilo o un grupo amino cuando se requiere una estrategia de desprotección ortogonal. Se usa también a veces para la formación cíclica de péptidos cuando el péptido está anclado a la resina por un grupo funcional de una cadena lateral. El grupo Alloc puede ser retirado usando tetrakis(trifenilfosfina)-paladio (0)en una disolución 37:2:1 de cloruro de metileno, ácido acético y N-metilmorfolina (NMM), durante dos horas. La elución de la resina debe levarse a cabo cuidadosamente con diisopropiletilamina (DIPEA) al 0,5% en DMF, 3x10 ml de dietiltiocarbamato al 0,5% en DMF y 5x10 ml de DCM:DMF 1:1.
Grupos protectores litográficos
Se usan en aplicaciones especiales como los microchips de proteínas. Estos grupos pueden ser retirados mediante la exposición a la luz solar.
Grupos activadores
Para el desanclaje del péptido es necesario activar primero el grupo carboxilo, importante paso para acelerar la reacción. Hay dos tipos principales de activadores: las carbodiimidas y los triazoles. Sin embargo, el uso de ésteres de pentafluorofenilo (FDPP, PFPOH)) y el BOP-Cl son útiles para la ciclación de péptidos.
Carbodiimidas

Estos reactivos fueron introducidos por Sheehan y Hess en 1955. Los más comunes son la diciclohexilcarbodiimida (DCC) y la disopropilcarbodiimida (DIC). Su reacción con un ácido carboxílico produce una O-acilurea muy reactiva. Durante la síntesis proteica artificial (como por ejemplo en sintetizadores Fmoc en fase sólida), normalmente se usa el extremo C-terminal como sitio de anclaje en que unir los aminoácidos.
Para aumentar la electrofilia del carboxilato, el oxígeno cargado negativamente tiene que ser activado previamente para convertirse en un grupo de partida más favorable. Para este propósito se emplea DCC. El oxígeno entonces actúa de forma nucleófila, atacando el carbono central de la DCC. La DCC se une temporalmente al carboxilato (ahora grupo éster) haciendo que se produzca de manera más eficaz el ataque nucleofílico por parte de un grupo amino, del aminoácido entrante, sobre el grupo carbonilo del extremo C-terminal.
El problema de las carbodiimidas es que son muy reactivas y pueden causar la racemización del aminoácido.
Triazoles
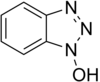
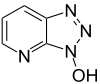
Se introdujeron en respuesta al problema de la racemización. Los más importantes son el 1-hidroxibenzotriazol (HOBt) y el 7-aza-1-hidroxibenzotriazol (HOAt), aunque se han desarrollado también otros. Estas sustancias reaccionan con la 0-acilurea y forman un éster activo menos reactivo y menos proclive a producir racemización. El HOAt es especialmente favorable por el efecto de grupo vecino. Recientemente se ha eliminado el HOBt de los catálogos comerciales químicos. Aunque se encuentra casi siempre en forma de hidrato, el HOBt puede ser explosivo cuando se lo deja deshidratar completamente, por lo que su transporte por tierra o aire está muy restringido.
Se han introducido alternativas al HOBt y el HOAt: una de las más económicas y con mayores posibilidades es el etilo 2-ciano 2-(hidroximino)acetato (con nombre comercial Oxyma Pure), que no es explosivo y cuya reactividad se encuentra entre la del HOBt y la del HOAt.
Los últimos avances han permitido eliminar totalmente las carbodiimidas. El éster activo es introducido en forma de sales de uronio o fosfonio de un anión no nucleofílico (tetrafluoroborato o hexafluorofosfato): HBTU, HATU, HCTU, PyBOP. Dos tipos de uronio del aditivo de acoplamiento del Oxyma Pure son los reactivos COMU y TOTU.
Formación regioselectiva de puentes disulfuro
Uno de los principales retos con el que todavía se enfrentan los métodos en fase sólida es la creación de múltiples enlaces disulfuro nativos. La combinación al azar de las cadenas da como resultado la formación de productos con puentes disulfuro no nativos.
La formación lineal de los enlaces disulfuro es el método de elección y se lleva a cabo mediante grupos protectores (GP) tiol. Diferentes GP tiol procuran distintos acercamientos en la protección ortogonal. Las cisteínas protegidas de forma ortogonal son incorporadas durante la fase sólida de la síntesis del péptido. La retirada sucesiva de estos GP permite la exposición de los grupos tiol libres y conduce a la formación de puentes disulfuro de forma lineal. El orden en que los GP se retiran es clave en la medida en que debe retirarse un solo grupo cada vez. En 1993, Kiso et al. fueron los primeros en informar sobre la síntesis total de la insulina siguiendo este método.
Los GP tiol deben poseer varias características: en primer lugar, el GP debe ser reversible en condiciones que no afecten a las cadenas laterales desprotegidas.; en segundo lugar, el GP debe poder resistir las condiciones de la SPPS; por último, la estrategia en la retirada de cada GP tiol debe ser tal que deje intactos otros GP tiol, si lo que se desea es un método ortogonal de protección. En otras palabras, la retirada del GP “a” no debe afectar al GP “b”.
Algunos de los GP tiol que se utilizan normalmente son el acetamidometilo (Acm), el terc-butilo (But), el 3-nitro-2-piridina-sulfenilo (NPYS), el 2-piridina-sulfenilo (Pyr) y el trifenilmetilo (tritilo o Trt). Debe tenerse en cuenta que el grupo NPYS puede reemplazar el grupo protector Acm para producir un tiol activo.
En la estrategia de formación lineal de puentes disulfuro que siguieron Kiso et al en la síntesis de la insulina, se sintetizó la cadena A siguiendo el siguiente esquema de protecciones: CysA6(But); CysA7(Acm); CysA11(But). De esta forma, el CysA20 queda desprotegido. La síntesis de la cadena B se llevó a cabo con la siguiente protección: CysB7(Acm) y CysB19(Pyr). El primer puente disulfuro, CysA20-CysB19, se formó mezclando las dos cadenas en urea 8 M. El pH 8 no afecta a los otros GP, como los grupos Acm o But.
De forma análoga, la formación del segundo puente disulfuro con iodina en ácido acético acuoso no afecta a los grupos But.
Una cuestión importante en la creación de los enlaces disulfuro es el orden en que se forman los S-S. En teoría, el orden en que los grupos tiol son expuestos para formar los puentes carece de relevancia, ya que las cisteínas están protegidas. Pero desde un punto de vista práctico, sin embargo, el orden en que se van formando los S-S puede influir sobremanera en el rendimiento. Esto puede deberse a que la formación del enlace CysA20-CysB19 puede colocar al grupo tiol de CysB7 muy próximo a CysA6 y CysA7, lo que resulta en la formación de múltiples productos disulfuro. Esto es una muestra más que pone de manifiesto el carácter de arte, además de ciencia, que posee la técnica de síntesis peptídica en fase sólida.
Síntesis de grandes péptidos
La elongación lineal, en que los aminoácidos se van conectando uno a uno, es un método ideal para péptidos pequeños de entre 2 y 100 residuos. Otro método es la condensación de fragmentos, en que lo que se acoplan son fragmentos de péptidos. Aunque el primer método puede elongar el péptido sin producir racemización, el rendimiento disminuye si se utiliza solo en la síntesis de péptidos grandes o muy polares. Por su parte, la condensación de fragmentos es preferible a la elongación gradual si se quieren sintetizar péptidos grandes y complejos, pero su uso debe limitarse si se quiere evitar la racemización.
La condensación de fragmentos no es muy recomendable ya que el fragmento acoplado puede resultar en gran exceso, lo cual puede ser una limitación dependiendo de la longitud del péptido.
El ligamiento químico es uno de los últimos avances que ha permitido la producción de péptidos de mayor tamaño. En este método, la cadena peptídicas desprotegidas reaccionan de forma quimioselectiva en medio acuoso. Un primer producto controlado cinéticamente se reorganiza para formar el enlace amida. En la forma más usual de ligamiento químico nativo se utiliza un péptido tioéster que reacciona con un residuo terminal de cisteína.
Eficacia de acoplamiento vs longitud del péptido
| Longitud del péptido | Eficacia de acoplamiento | Coupling Efficiency | Coupling Efficiency | Coupling Efficiency | Coupling Efficiency |
| 0 | 0,995 | 0,99 | 0,98 | 0,97 | 0,96 |
| 5 | 0,98 | 0,95 | 0,92 | 0,89 | 0,85 |
| 10 | 0,96 | 0,91 | 0,83 | 0,76 | 0,69 |
| 15 | 0,93 | 0,87 | 0,75 | 0,65 | 0,56 |
| 20 | 0,91 | 0,83 | 0,68 | 0,56 | 0,46 |
| 25 | 0,89 | 0,79 | 0,62 | 0,48 | 0,38 |
| 30 | 0,86 | 0,75 | 0,56 | 0,41 | 0,31 |
| 35 | 0,84 | 0,71 | 0,50 | 0,36 | 0,25 |
| 40 | 0,82 | 0,67 | 0,45 | 0,30 | 0,20 |
| 45 | 0,80 | 0,63 | 0,41 | 0,26 | 0,17 |
| 50 | 0,78 | 0,60 | 0,37 | 0,22 | 0,14 |
| 55 | 0,76 | 0,58 | 0,34 | 0,19 | 0,11 |
| 60 | 0,74 | 0,55 | 0,30 | 0,17 | 0,09 |
| 65 | 0,73 | 0,53 | 0,27 | 0,14 | 0,07 |
| 70 | 0,71 | 0,50 | 0,25 | 0,12 | 0,06 |
Síntesis de péptidos asistida por microondas
Si bien la radiación de microondas se ha usado desde finales de los años 40, no fue hasta 1986 cuando la energía microondas se utilizó en química orgánica. Durante los años 80 y 90 del siglo pasado esta energía ayudó a acelerar reacciones químicas que de otro modo hubieran tardado horas o incluso días es completarse.
Desde finales de los 90 a principios de 2000, tuvieron lugar una serie de mejoras técnicas que permitieron que los sintetizadores de microondas produjesen paquetes de diferentes energías, de forma que la temperatura de la reacción pudo controlarse.
La radiación de microondas que su usa en la síntesis de péptidos posee una sola frecuencia y permite una mayor penetración de la muestra, al contrario de los electrodomésticos de microondas que conocemos.
En la síntesis de péptidos, la radiación de microondas se ha usado para la compleción de secuencias peptídicas de gran longitud con rendimientos muy elevados y poco grado de racemización. El acoplamiento de los aminoácidos a la cadena peptídica no se cataliza solo mediante el aumento de la temperatura sino también debido a la radiación electromagnética alterna con la cual se alinea continuamente el esqueleto polar del polipéptido. A causa de este fenómeno, la energía microondas puede evitar la agregación y por tanto incrementar el rendimiento final del péptido.
Pese a todo, no hay pruebas de que el uso de las microondas deba preferirse al simple calentamiento y algunos laboratorios contemplan esta técnica simplemente como una manera más rápida de calentar la peptidilresina. El calentamiento por encima de los 50oC o 55oC evita también la agregación y acelera el acoplamiento.
Pese a las obvias ventajas de la radiación de microondas en la síntesis de péptidos, la principal desventaja es la racemización que puede producirse durante el acoplamiento de la cisteína y la histidina. La reacción de acoplamiento de estos aminoácidos se realiza a temperaturas más bajas que los otros 18 aminoácidos naturales. Por otra parte, hay un grupo de aminoácidos que no sobrevive a las microondas o al calor en general. Uno de los efectos no deseados más importantes es la deshidratación (pérdida de agua) que puede ser muy elevada en ciertos péptidos, como el polipéptido pancreático (PP), efecto secundario que puede también observarse con el simple calentamiento.
Hasta enero de 2009 se trabajaba con más de 200 sintetizadores de péptidos con microondas, y su uso no hace sino aumentar.
- Atherton, E.; Sheppard, R.C. (1989). Solid Phase peptide synthesis: a practical approach. Oxford, England: IRL Press. ISBN 0199630674.
- Stewart, J.M.; Young, J.D. (1984). Solid phase peptide synthesis (2nd edición). Rockford: Pierce Chemical Company. p. 91. ISBN 0935940030.
| Control de autoridades |
|
|---|
-
 Datos: Q110820614
Datos: Q110820614