
| Síndrome mielodisplásico | ||
|---|---|---|
| Especialidad | hematología | |

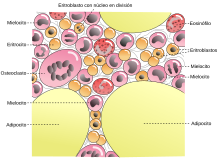

Los síndromes mielodisplásicos o anemia refractaria son un grupo de neoplasias hematológicas caracterizadas por diferenciación anormal, dismorfología y citopenias producidas por apoptosis exagerada de precursores hematopoyéticos en la médula ósea, anormalidades cromosómicas, mutaciones genéticas somáticas; que, en general, tienen tendencia a evolucionar a leucemia mieloide aguda. Ocurren con mayor frecuencia en personas mayores, pero también pueden acontecer en los jóvenes.
Por lo general no se observan síntomas inicialmente. Posteriormente, se pueden presentar síntomas como sensación de cansancio, dificultad para respirar, trastornos hemorrágicos, anemia o infecciones frecuentes.
Los factores de riesgo incluyen quimioterapia o radioterapia previa, exposición a ciertas sustancias químicas como humo del tabaco, pesticidas y benceno, y exposición a metales pesados como el mercurio o el plomo. Los problemas con la formación de glóbulos resultan en una combinación de recuentos bajos de glóbulos rojos, plaquetas y glóbulos blancos. Algunos tipos tienen un aumento de células sanguíneas inmaduras, llamadas blastos, en la médula ósea o en la sangre. Los tipos de este síndrome se basan en cambios específicos en las células sanguíneas y la médula ósea.
Los tratamientos pueden incluir atención de apoyo, terapia con medicamentos y trasplante de células madre hematopoyéticas. La atención de apoyo puede incluir transfusiones de sangre, medicamentos para aumentar la producción de glóbulos rojos y antibióticos. La terapia con medicamentos puede incluir los medicamentos lenalidomida, globulina antitimocítica y azacitidina.
Aproximadamente siete de cada 100 000 personas se ven afectadas, y aproximadamente cuatro de cada 100 000 personas adquieren la afección cada año. La edad típica de inicio es de 70 años. El pronóstico depende del tipo de células afectadas, la cantidad de blastos en la médula ósea o en la sangre y los cambios presentes en los cromosomas de las células afectadas. El tiempo de supervivencia típico después del diagnóstico es de 2,5 años.
Las condiciones del síndrome se reconocieron por primera vez a inicios del siglo XX. El nombre actual entró en uso en 1976.
Signos y síntomas
El signo más común es la anemia, lo que significa que no hay suficientes glóbulos rojos maduros para transportar oxígeno. Ante la presencia de anemia, infecciones o hemorragia, se debe solicitar un análisis de sangre para obtener un recuento de cada clase de glóbulos en la sangre en el que se pueden o no apreciar formas eritroblásticas, pero siempre menor al 20 % ya que un porcentaje mayor constituiría un diagnóstico de leucemia. Si los resultados de los análisis de sangre no son normales, el médico podría hacer una biopsia de médula ósea para determinar el tipo de enfermedad que se padece y planificar el mejor tratamiento.
También es posible que no haya suficientes glóbulos blancos en la sangre para combatir las infecciones. Si el número de las plaquetas de la sangre está por debajo de lo normal, la persona puede sangrar o sufrir la aparición de hematomas más fácilmente de lo habitual.
Es posible desarrollarlo después de recibir un tratamiento con medicamento o radioterapia para otras enfermedades, o sin que haya una causa conocida. Los síndromes mielodisplásicos pueden convertirse en leucemia mieloide aguda, un tipo de enfermedad maligna en la que se producen demasiados glóbulos blancos.
Muchas personas son asintomáticas y la citopenia sanguínea u otros problemas se identifican como parte de un hemograma de rutina.
Aunque existe cierto riesgo de desarrollar leucemia mieloide aguda, alrededor del 50 % de las muertes se producen como resultado de una hemorragia o una infección. Sin embargo, la leucemia que se produce como resultado de la mielodisplasia es notoriamente resistente al tratamiento. La mayoría de los pacientes sintomáticos se quejan de la aparición gradual de fatiga y debilidad, disnea y palidez, pero al menos la mitad de los pacientes son asintomáticos y su afección se descubre solo de manera incidental en los hemogramas de rutina. La exposición previa a la quimioterapia o la radiación es un factor importante en el historial médico de la persona. La fiebre, la pérdida de peso y la esplenomegalia deberían indicar una neoplasia mielodisplásica/mieloproliferativa en lugar de un proceso mielodisplásico puro.
Diagnóstico
El diagnóstico suele completarse por medio de:
- Hemograma completo y examen del frotis de sangre: la morfología del frotis de sangre puede proporcionar pistas sobre la anemia hemolítica, acumulación de plaquetas que conduce a una trombocitopenia espuria o leucemia.
- Exámenes de sangre para eliminar otras causas comunes de citopenias como lupus, hepatitis, vitamina B12, ácido fólico u otras deficiencias vitamínicas, insuficiencia renal o cardíaca, VIH, anemia hemolítica y gammapatía monoclonal
Se debe considerar la detección de cáncer mediante:
- Examen de médula ósea por un hematopatólogo, esto es necesario para establecer el diagnóstico, ya que todos los hematopatólogos consideran que la médula displásica es la característica clave de la mielodisplasia.
- Estudios citogenéticos o cromosómicos: idealmente se realizan en el aspirado de médula ósea. La citogenética convencional requiere una muestra fresca ya que se induce a las células vivas a entrar en metafase para permitir que se vean los cromosomas.
- Las pruebas de hibridación fluorescente in situ interfase, generalmente solicitadas junto con las pruebas citogenéticas convencionales, ofrecen una detección rápida de varias anomalías cromosómicas asociadas con el síndrome, incluidas del 5q, −7, +8 y del 20q.
- No se deben pasar por alto las pruebas de deficiencia de cobre, ya que morfológicamente puede parecerse al SMD en biopsias de médula ósea.
Clasificación
Los síndromes mielodisplásicos se agrupan dependiendo de la apariencia que tengan las células de la médula ósea y los glóbulos bajo el microscopio. La clasificación de la OMS es:
- Citopenia refractaria con displasia unilínea.
- Anemia refractaria con sideroblastos en anillo.
- Citopenia refractaria con displasia multilínea.
- Anemia refractaria con exceso de blastos-1 (AREB-1) {5 a 9 % blastos en ESP}.
- Anemia refractaria con exceso de blastos-2 (AREB-2) {10 a 19 % blastos en ESP}.
- Síndrome mielodisplásico-inclasificable.
- SMD asociado con deleción(5q) aislada.
- SMD de la infancia.
Anatomía patológica
El síndrome mielodisplásico no permite la maduración de la médula ósea, es decir, la médula ósea nunca madura como una médula ósea normal. Hace también que el corazón trabaje más de lo normal. El rasgo más característico, quizá, es la diferenciación alterada (displásica) que afecta a todos los linajes no linfoides (eritroide, granulocítico, monocítico y megacariocítico).
Dentro de la serie eritroide, las anomalías comunes comprenden sideroblastos en anillo (anulares), eritroblastos con mitocondrias cargadas de hierro, visibles como gránulos perinucleares en los aspirados o biopsias teñidos con azul de prusia; maduración megaloblástica que recuerda a la observada en la deficiencia de vitamina B12 y folato, y anomalías de la maduración nuclear, que se reconoce como núcleos con contornos mal formados, con frecuencia polipoides.
Los neutrófilos contienen muchas veces un número disminuido de gránulos secundarios, granulaciones tóxicas o cuerpos de Döhle. Son frecuentes las seudo-células de Pelger-Hüet, neutrófilos con solo dos lóbulos nucleares e incluso se pueden encontrar neutrófilos que carecen por completo de segmentación nuclear. También son característicos los megacariocitos con lóbulos nucleares únicos o con múltiples núcleos separados, ("pawn ball megakaryocytes").
Los mieloblastos pueden estar aumentados, pero constituyen menos de 20 % de la celularidad global. La sangre periférica contiene con frecuencia seudo-células de Pelger-Hüet, plaquetas gigantes, macrocitos, poiquilocitos y una monocitosis relativa o absoluta.
Pronóstico
El SMD primario afecta principalmente a individuos de más de 60 años de edad y es el primer diagnóstico a descartar de anemia macrocítica en este grupo. En los casos sintomáticos se presenta con debilidad, infecciones y hemorragia debidas a la pancitopenia. En hasta la mitad de los casos, el SMD se descubre de modo incidental en examen de sangre solicitado por otro motivo.
Enlaces externos
- Esta obra contiene una traducción derivada de «Myelodysplastic syndrome» de Wikipedia en inglés, concretamente de esta versión, publicada por sus editores bajo la Licencia de documentación libre de GNU y la Licencia Creative Commons Atribución-CompartirIgual 3.0 Unported.
| Control de autoridades |
|
|---|
-
 Datos: Q954625
Datos: Q954625
-
 Multimedia: Myelodysplastic syndrome / Q954625
Multimedia: Myelodysplastic syndrome / Q954625