
La heterogeneidad tumoral describe la observación de que diferentes células tumorales pueden mostrar distintos perfiles morfológicos y fenotípicos, que incluyen morfología celular, expresión génica, metabolismo, motilidad, proliferación y potencial metastásico. Este fenómeno ocurre tanto entre tumores (heterogeneidad intertumoral) como dentro de los tumores (heterogeneidad intratumoral). Un nivel mínimo de heterogeneidad intratumoral es una simple consecuencia de la imperfección de la replicación del ADN: siempre que una célula (normal o cancerosa) se divide, se adquieren algunas mutaciones conduce a una población diversa de células cancerosas. La heterogeneidad de las células cancerosas presenta importantes desafíos en el diseño de estrategias de tratamiento eficaces. Sin embargo, la investigación para comprender y caracterizar la heterogeneidad puede permitir una mejor comprensión de las causas y la progresión de la enfermedad. A su vez, esto tiene el potencial de guiar la creación de estrategias de tratamiento más refinadas que incorporen el conocimiento de la heterogeneidad para producir una mayor eficacia.
Se ha observado heterogeneidad tumoral en leucemias, mama, próstata, colon, cerebro, esófago, cabeza y cuello, vejiga y carcinomas ginecológicos, liposarcoma, y mieloma múltiple.
Modelos de heterogeneidad
Se utilizan dos modelos para explicar la heterogeneidad de las células tumorales. Estos son el modelo de células madre cancerosas y el modelo de evolución clonal. Los modelos no son mutuamente excluyentes y se cree que ambos contribuyen a la heterogeneidad en cantidades variables en los diferentes tipos de tumores.
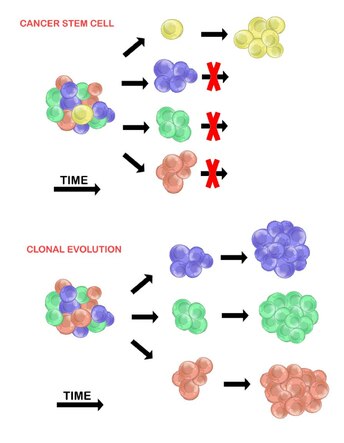
Células madre cancerosas
El modelo de células madre cancerosas afirma que dentro de una población de células tumorales, solo hay un pequeño subconjunto de células que son tumorigénicas (capaces de formar tumores). Estas células se denominan células madre cancerosas (CSC o cancer stem cells) y están marcadas por la capacidad tanto de autorrenovarse como de diferenciarse en una progenie no tumorigénica. El modelo CSC postula que la heterogeneidad observada entre las células tumorales es el resultado de diferencias en las células madre de las que se originaron. La variabilidad de las células madre a menudo es causada por cambios epigenéticos, pero también puede resultar de la evolución clonal de la población de CSC donde pueden acumularse mutaciones genéticas ventajosas en las CSC y su progenie.
Se ha demostrado evidencia del modelo de células madre cancerosas en múltiples tipos de tumores, como leucemias, glioblastoma, cáncer de mama, y cáncer de próstata.
Sin embargo, la existencia de CSC todavía está en debate. Una razón de esto es que los marcadores de CSC han sido difíciles de reproducir en múltiples tumores. Además, los métodos para determinar el potencial tumorigénico utilizan modelos de xenoinjerto. Estos métodos adolecen de limitaciones inherentes tales como la necesidad de controlar la respuesta inmunitaria en el animal trasplantado y la diferencia significativa en las condiciones ambientales desde el sitio del tumor primario hasta el sitio del xenoinjerto (por ejemplo, ausencia de las moléculas exógenas o cofactores requeridos). Esto ha provocado algunas dudas sobre la precisión de los resultados de CSC y las conclusiones sobre qué células tienen potencial tumorigénico.
Evolución clonal
El modelo de evolución clonal fue propuesto por primera vez en 1976 por Peter Nowell. En este modelo, los tumores surgen de una sola célula mutada, acumulando mutaciones adicionales a medida que avanza. Estos cambios dan lugar a subpoblaciones adicionales, y cada una de estas subpoblaciones tiene la capacidad de dividirse y mutar aún más. Esta heterogeneidad puede dar lugar a subclones que posean una ventaja evolutiva sobre los demás dentro del entorno del tumor, y estos subclones pueden llegar a ser dominantes en el tumor con el tiempo. Cuando se propuso, este modelo permitió comprender el crecimiento tumoral, el fracaso del tratamiento y la agresión tumoral que se produce durante el proceso natural de formación del tumor.
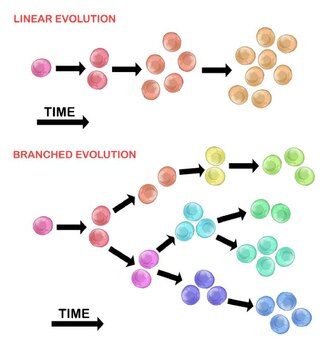
La evolución de la célula tumoral inicial puede ocurrir por dos métodos:
Expansión lineal
Las mutaciones ordenadas secuencialmente se acumulan en genes impulsores, genes supresores de tumores y enzimas reparadoras del ADN, lo que resulta en la expansión clonal de las células tumorales. Es menos probable que la expansión lineal refleje el criterio de valoración de un tumor maligno porque la acumulación de mutaciones es estocástica en los tumores heterogéneos.
Expansión ramificada
La expansión a múltiples poblaciones subclonales se produce a través de un mecanismo de división. Este método está más asociado con la heterogeneidad tumoral que con la expansión lineal. La adquisición de mutaciones es aleatoria como resultado de una mayor inestabilidad genómica con cada generación sucesiva. La acumulación mutacional a largo plazo puede proporcionar una ventaja selectiva durante ciertas etapas de la progresión del tumor. El microambiente del tumor también puede contribuir a la expansión del tumor, ya que es capaz de alterar las presiones selectivas a las que están expuestas las células tumorales.
Tipos y causas de heterogeneidad
Se han observado múltiples tipos de heterogeneidad entre las células tumorales, derivados de la variabilidad tanto genética como no genética.
Heterogeneidad genética
La heterogeneidad genética es una característica común de los genomas tumorales y puede surgir de múltiples fuentes. Algunos cánceres se inician cuando factores exógenos introducen mutaciones, como la radiación ultravioleta (cánceres de piel) y el tabaco (cáncer de pulmón). Una fuente más común es la inestabilidad genómica, que a menudo surge cuando las vías reguladoras clave se interrumpen en las células. Algunos ejemplos incluyen mecanismos de reparación de ADN alterados que pueden conducir a un aumento de errores de replicación y defectos en la maquinaria de la mitosis que permiten la ganancia o pérdida a gran escala de cromosomas completos. Además, es posible que la variabilidad genética aumente aún más con algunas terapias contra el cáncer (tratamiento con temozolomida y otros fármacos de quimioterapia).
La heterogeneidad tumoral mutacional se refiere a variaciones en la frecuencia de mutación en diferentes genes y muestras y puede ser explorada por MutSig Archivado el 3 de octubre de 2017 en Wayback Machine.. La etiología de los procesos mutacionales puede variar considerablemente entre muestras tumorales del mismo o diferentes tipos de cáncer y puede manifestarse en diferentes perfiles mutacionales dependientes del contexto. Puede ser explorado por firmas mutacionales COSMIC o MutaGene.
Otra heterogeneidad
Las células tumorales también pueden mostrar heterogeneidad entre sus perfiles de expresión. Esto a menudo es causado por cambios epigenéticos subyacentes. Se han detectado variaciones en las firmas de expresión en diferentes regiones de muestras tumorales dentro de un individuo. Los investigadores han demostrado que las mutaciones que afectan convergentes H3K36 metiltransferasa SETD2 y la histona H3K4 desmetilasa KDM5C surgió en secciones tumorales espacialmente separadas. De manera similar, MTOR, un gen que codifica una quinasa reguladora celular, ha demostrado ser constitutivamente activo, aumentando así la fosforilación de S6. Esta fosforilación activa puede servir como biomarcador en el carcinoma de células claras.
La heterogeneidad mecanoquímica es un sello distintivo de las células eucariotas vivas. Tiene un impacto en la regulación de genes epigenéticos. Los procesos mecanoquímicos dinámicos heterogéneos regulan las interrelaciones dentro del grupo de superficies celulares a través de la adhesión. El desarrollo y la propagación del tumor se acompaña de un cambio en la dinámica caótica heterogénea del proceso de interacción mecanoquímica en las células del grupo, incluidas las células dentro del tumor, y es jerárquico para el anfitrión de pacientes con cáncer. Los fenómenos biológicos de heterogeneidad mecanoquímica pueden utilizarse para el diagnóstico diferencial del cáncer gástrico contra pacientes con inflamación de la mucosa gástrica y para aumentar la actividad antimetastásica de las células dendríticas basadas en vacunas cuando se utilizan micropartículas de células tumorales heterogeneizadas mecánicamente para su carga. Existe también un posible abordaje metódico basado en las técnicas diagnósticas y terapéuticas por imagen ecográfica simultánea, respecto al efecto mecanoquímico sobre conglomerados de nanoburbujas con fármacos en el tumor.
Microambiente tumoral
La heterogeneidad entre las células tumorales puede aumentar aún más debido a la heterogeneidad en el microambiente del tumor. Las diferencias regionales en el tumor (por ejemplo, la disponibilidad de oxígeno) imponen diferentes presiones selectivas sobre las células tumorales, lo que conduce a un espectro más amplio de subclones dominantes en diferentes regiones espaciales del tumor. La influencia del microambiente en la dominancia clonal también es una razón probable de la heterogeneidad entre los tumores primarios y metastásicos observada en muchos pacientes, así como la heterogeneidad inter tumoral observada entre pacientes con el mismo tipo de tumor.
Implicaciones y desafíos
Resistencia al tratamiento
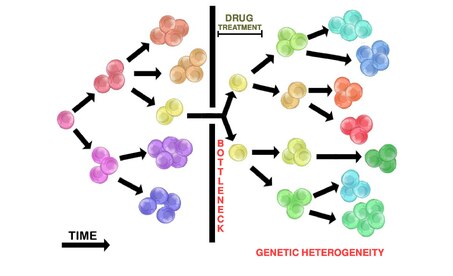
Los tumores heterogéneos pueden exhibir diferentes sensibilidades a los fármacos citotóxicos entre diferentes poblaciones clonales. Esto se atribuye a interacciones clonales que pueden inhibir o alterar la eficacia terapéutica, lo que plantea un desafío para terapias exitosas en tumores heterogéneos (y sus metástasis heterogéneas).
La administración de fármacos en tumores heterogéneos rara vez destruirá todas las células tumorales. La población tumoral heterogénea inicial puede producir un cuello de botella, de modo que sobrevivirán pocas células resistentes a los fármacos (si las hay). Esto permite que las poblaciones de tumores resistentes se repliquen y desarrollen un nuevo tumor a través del mecanismo de evolución ramificada (ver más arriba). El tumor repoblado resultante es heterogéneo y resistente al tratamiento farmacológico inicial utilizado. El tumor repoblado también puede regresar de una manera más agresiva.
La administración de fármacos citotóxicos a menudo da como resultado una reducción inicial del tumor. Esto representa la destrucción de poblaciones subclonales iniciales no resistentes dentro de un tumor heterogéneo, dejando solo clones resistentes. Estos clones resistentes ahora contienen una ventaja selectiva y pueden replicarse para repoblar el tumor. La replicación probablemente ocurrirá a través de la evolución ramificada, lo que contribuirá a la heterogeneidad del tumor. El tumor repoblado puede parecer más agresivo. Esto se atribuye a la ventaja selectiva de las células tumorales resistentes a los fármacos.
Descubrimiento de biomarcadores
Debido a las diferencias genéticas dentro y entre los tumores, los biomarcadores que pueden predecir la respuesta al tratamiento o el pronóstico pueden no ser de aplicación generalizada. Sin embargo, se ha sugerido que el nivel de heterogeneidad en sí mismo puede usarse como un biomarcador, ya que es más probable que los tumores más heterogéneos contengan subclones resistentes al tratamiento. Todavía se están realizando más investigaciones sobre el desarrollo de biomarcadores que expliquen la heterogeneidad.
Sistemas modelo
Los sistemas de modelos actuales generalmente carecen de la heterogeneidad que se observa en los cánceres humanos. Para estudiar con precisión la heterogeneidad tumoral, debemos desarrollar modelos preclínicos más precisos. Uno de estos modelos, el xenoinjerto de tumor derivado del paciente, ha demostrado una excelente utilidad para preservar la heterogeneidad del tumor al tiempo que permite un estudio detallado de los impulsores de la aptitud clonal. Sin embargo, incluso este modelo no puede captar toda la complejidad del cáncer.
Estrategias actuales
Si bien el problema de identificar, caracterizar y tratar la heterogeneidad tumoral aún se encuentra bajo investigación activa, se han propuesto algunas estrategias efectivas, que incluyen soluciones tanto experimentales como computacionales.
Experimental
-
Enfoque enfocado: analizar un locus genético específico o un conjunto de loci. Esto puede ocurrir mediante la detección de desequilibrios alélicos (el ADN tumoral se compara con el ADN de la línea germinal), la amplificación de regiones cromosómicas (FISH) y/o la secuenciación de genes específicos. Este método se utiliza para rastrear la evolución de una mutación específica de interés o para confirmar una mutación que los investigadores pueden sospechar en un tumor.
- Ventajas: permite el análisis de genes específicos (genes impulsores, supresores de tumores). El proceso es simple con una interpretación directa de los resultados. La FISH y la inmunofluorescencia permiten centrarse en los subtipos de células tumorales.
- Desventajas: un análisis limitado pasará por alto mutaciones importantes adicionales y patrones de expansión clonal. Los desequilibrios alélicos pueden ser difíciles de verificar utilizando marcadores de microsatélites, por lo que requieren verificación mediante una técnica independiente. La FISH requiere una gran cantidad de células y requiere mucha mano de obra.
-
Enfoque de todo el genoma: análisis de todo el genoma en muestras de tumores. Esto se puede hacer mediante cariotipo o hibridación genómica comparativa (CGH) para detectar anomalías cromosómicas. La secuenciación de biopsias tumorales es cada vez más común.
- Ventajas: No se basa en conocimientos previos para identificar variantes. El cariotipo identifica regiones de grandes anomalías cromosómicas. CGH proporciona una cobertura imparcial y permite detectar desequilibrios alélicos a pequeña escala (matrices SNP). La secuenciación identificará cualquier variante que contribuya a la heterogeneidad del tumor.
- Desventajas: es difícil determinar el impacto funcional de las variantes (neutrales o patógenas). Resolución limitada. El cariotipo de células cultivadas puede estar sesgado hacia el crecimiento preferencial de subpoblaciones de células tumorales seleccionadas. Resolución limitada en ambos métodos. El enfoque del genoma completo puede generar grandes cantidades de datos y ser difícil de interpretar.
- Estrategia de muestreo multirregional: generalmente requiere múltiples muestras tumorales posquirúrgicas de regiones separadas de un tumor microdisecado. Es importante evitar la contaminación de células no malignas para garantizar una representación precisa de la expresión génica y la composición genética observada únicamente dentro de las células tumorales. El análisis del ADN tumoral dentro de las regiones espacialmente separadas permite la construcción de un modelo evolutivo tridimensional de heterogeneidad tumoral. El muestreo multirregional se utiliza a menudo en combinación con el enfoque de todo el genoma para establecer este modelo de expansión de heterogeneidad 3D.
- Muestreo longitudinal: a través de la progresión del tumor o la progresión del tratamiento, en algunos casos se ha utilizado la obtención de muestras tumorales en múltiples momentos. Esto se ha sugerido como un método confiable para rastrear la evolución clonal. Sin embargo, esta técnica resulta desafiante en la práctica porque requiere biopsia invasiva periódica. Una nueva investigación sobre la utilización de ADN tumoral libre de células circulantes en la sangre puede proporcionar una forma no invasiva de identificar biomarcadores durante el tratamiento. El muestreo longitudinal utilizado en combinación con el enfoque de todo el genoma permitirá la identificación de las mutaciones acumuladas de células tumorales a lo largo del tiempo. Esto, a su vez, puede identificar las mutaciones impulsoras clave (observadas en las muestras iniciales de tumores).
- La terapia adaptativa puede usarse para prevenir un mayor crecimiento tumoral ajustando la dosis del fármaco y el momento de administración del fármaco en función de la respuesta del tumor. Se supone que esta estrategia evita que las variantes resistentes dominen un tumor. Sin embargo, se requiere más investigación sobre su aplicabilidad.
Secuenciación
- Se puede utilizar la secuenciación de tumores a granel, donde el ADN se extrae de una mezcla de células tumorales y se analiza de una vez. La presencia de poblaciones de tumores heterogéneas (subclones) introduce desafíos adicionales como:
- La incapacidad para detectar mutaciones en subclones raros. Dado que estas mutaciones ocurrirán con baja frecuencia en la muestra combinada, pueden ser indistinguibles del ruido de fondo. Sin embargo, se están desarrollando activamente muchas variantes de llamadas que están diseñadas específicamente para datos de cáncer y tienen como objetivo identificar variantes raras presentes en poblaciones subclonales más pequeñas. Por lo general, estos utilizan ADN normal emparejado como un medio para distinguir la variación somática verdadera de la variación de la línea germinal y el error de secuenciación de fondo.
- La incapacidad para determinar qué subclones contienen cada mutación. Dado que los datos se agrupan, no está claro qué mutaciones coexisten y de qué poblaciones se originan. Se están desarrollando nuevas herramientas que intentan resolver la estructura clonal utilizando frecuencias alélicas para las mutaciones observadas.
-
La secuenciación unicelular es una técnica nueva que es valiosa para evaluar la heterogeneidad tumoral porque puede caracterizar células tumorales individuales. Esto significa que el perfil mutacional completo de múltiples células distintas se puede determinar sin ambigüedad. Si bien con la tecnología actual, es difícil evaluar un número suficientemente grande de células individuales para obtener poder estadístico, los datos de tumores unicelulares tienen múltiples ventajas, que incluyen:
- La capacidad de construir un árbol filogenético que muestre la evolución de las poblaciones tumorales. Usando secuencias de genoma completo o pseudo-secuencias basadas en SNP de células individuales, se puede estimar la evolución de los subclones. Esto permite la identificación de poblaciones que han persistido a lo largo del tiempo y puede reducir la lista de mutaciones que potencialmente confieren una ventaja de crecimiento o resistencia al tratamiento en subclones específicos. Los algoritmos para inferir una filogenia tumoral a partir de datos de secuenciación de ADN de una sola célula incluyen SCITE, OncoNEM, SiFit, SiCloneFit, PhISCS, y PhISCS-BnB.
- La secuenciación de secciones se puede realizar en múltiples porciones de un solo tumor sólido, y la variación en las frecuencias de mutación entre las secciones se puede analizar para inferir la estructura clonal. Las ventajas de este enfoque sobre la secuenciación única incluyen más poder estadístico y disponibilidad de información más precisa sobre el posicionamiento espacial de las muestras. Este último puede usarse para inferir la frecuencia de clones en secciones y proporcionar información sobre cómo evoluciona un tumor en el espacio. Para inferir los genotipos de los clones y los árboles filogenéticos que modelan la evolución de un tumor en el tiempo, se desarrollaron varios métodos computacionales incluyendo Clomial, cloneHD, PhyloWGS, PyClone, Cloe, phyC, Canopy, TargetClone, ddClone, PASTRI, GLClone, TRaIT, WSCUnmix, B-SCITE., ThetA, SIFA, Sclust, SeqClone, CALDER, BAMSE, Meltos, SubMARine, y RNDCLONE.
Véase también
| Control de autoridades |
|
|---|
-
 Datos: Q15917291
Datos: Q15917291